


Pharmacophor-geführtes virtuelles Screening zur Wiederverwendung von Medikamenten
Die Neuverwendung von Arzneimitteln ist eine seit langem etablierte Strategie zur Identifizierung neuer Anwendungen für zugelassene oder in der Erprobung befindliche Therapeutika, die über die ursprüngliche medizinische Indikation hinausgehen. 1 Seit der zufälligen Neupositionierung eines Medikaments, das gegen Bluthochdruck und Angina pectoris entwickelt wurde, zu Pfizers berüchtigter „kleiner blauer Pille“, allgemein als Viagra bezeichnet, ist die Neubestimmung von Medikamenten in der Öffentlichkeit nicht mehr so stark in den Vordergrund gerückt. Die weltweite Dringlichkeit von Therapeutika zur Behandlung von COVID-19 hat dieser Medikamentenstrategie erneut die Aufmerksamkeit der Massen verschafft.
Die 3D-Proteinmodellierung umfasst mehrere molekulare Modellierungs- und Simulationsmethoden für die Wiederverwendung von Medikamenten. In einem früheren Beitrag dieser Reihe wurde das molekulare Docking als Methode zur Identifizierung möglicher Inhibitoren untersucht, die an die SARS-CoV-2-Hauptprotease binden. Kann eine Alternative in silico Verfahren wie das Pharmakophor-Modelling vergleichbare Ergebnisse erzielen und zusätzliche Beweise liefern, die die Auswahl eines Kandidaten gegenüber einem anderen belegen?
Eine alternative Route nehmen
Die Modellierung von Pharmakophoren liefert eine Abstraktion der molekularen Merkmale, die für die Erkennung eines Liganden durch ein Proteinziel erforderlich sind. Seine Darstellung molekularer Wechselwirkungen und Bindungen bietet eine kontrastierende Perspektive zu klassischen Simulationsmethoden.
Wir haben einen ersten Datensatz mehrerer SARS-CoV-2-Hauptprotease-Proteinstrukturen von Diamond Light Source erhalten. 2 Wir haben diese Strukturen ausgerichtet und dann das im BIOVIA Discovery Studio verfügbare Protokoll „Interaction Pharmacophore Generation“ verwendet, um Pharmakophore zu generieren, die die Nicht-Bindungs-Wechselwirkungen jedes Rezeptor-Ligand-Komplexes darstellen. Die Gesamtzahl der Merkmale in den Pharmakophoren reichte von zwei Merkmalen für die Kristallstrukturen 5R80 und 5R7Y bis zu einem Modell mit neun Merkmalen für 6LU7. Wir haben die Pharmakophore der einzelnen Komplexe zu einem einzigen Modell zusammengeführt und eng geclusterte Merkmale bearbeitet. Das endgültige Modell umfasste 14 Merkmale, die intermolekulare Kontakte zwischen der Protease und möglichen niedermolekularen Bindern darstellen.
Anschließend haben wir in silico gespielt Alanin-Scanning-Mutagenese an den Resten des aktiven Zentrums aller Komplexe, um zu identifizieren, welche Reste die Bindungsaffinität (Hotspot) dieses Protein-Ligand-Komplexes verringerten, wenn er mutiert wurde. Von allen Komplexen identifizierten wir acht Reste (HIS41, MET49, ASN142, HIS163, MET165, PRO168, GLN189 und GLN192) als Hotspots in mindestens einem Komplex und drei Reste, die Hotspots in mindestens vier Komplexen waren. Im 14-Merkmal-Pharmakophormodell korrespondierten sechs Merkmale mit Wechselwirkungen mit einem der acht Hotspot-Reste. Ein zweiter Datensatz von nicht-kovalentem M PRO Ligandenkomplexe, die von der Diamond Light Source 2 freigesetzt werden enthüllten eine Reihe verschiedener Bindungsmodi, was uns dazu veranlasste, die sechs Schlüsselmerkmale der Pharmakophore in zwei Gruppen zu unterteilen, die im nächsten Schritt des virtuellen Screenings verwendet werden.
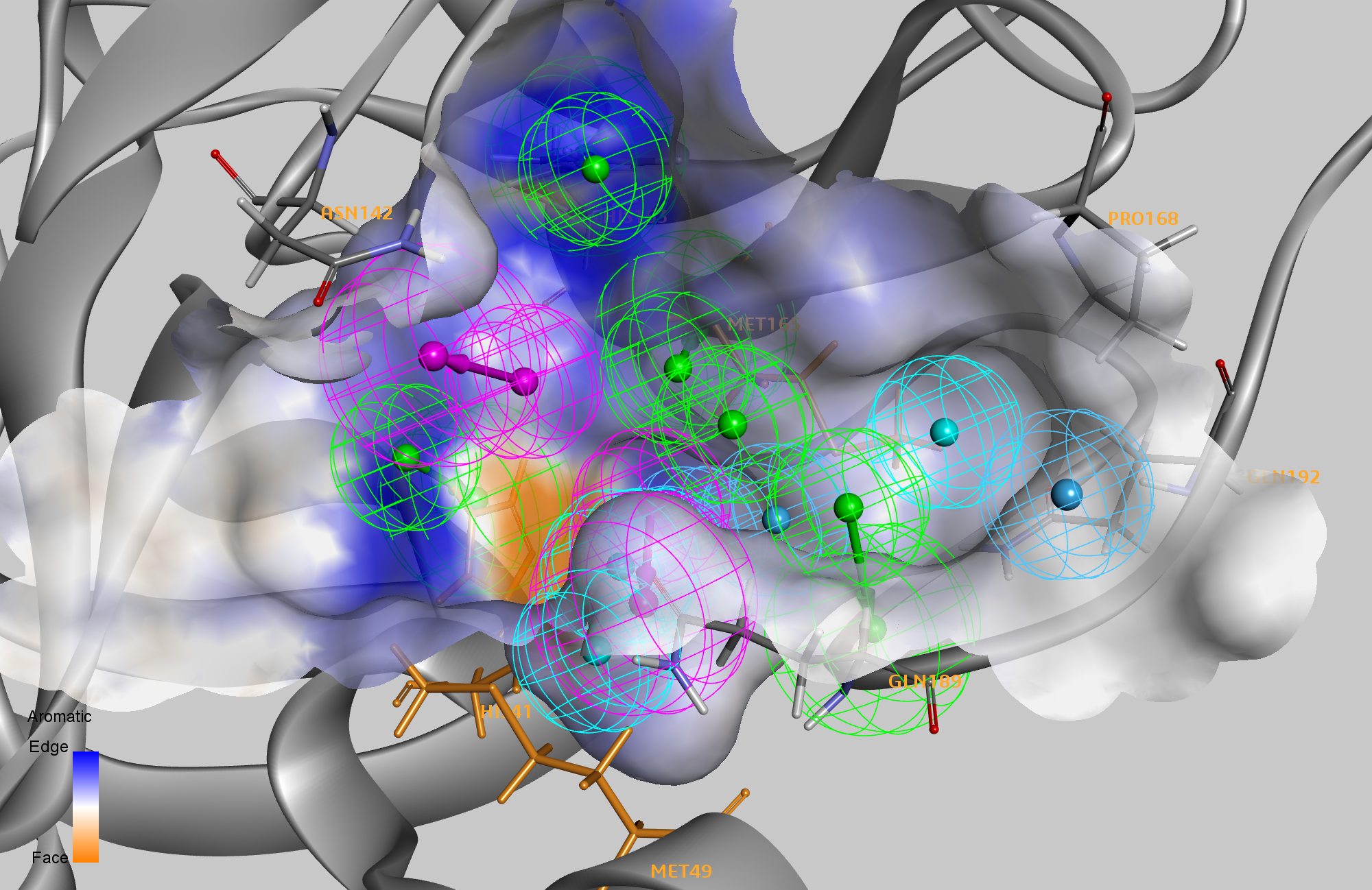
Wir haben dieses Pharmakophor-Modell nun verwendet, um ein virtuelles Screening einer ausgewählten Bibliothek durchzuführen, die 2.650 von der FDA zugelassene Medikamente enthält, die mehrere Ligandenbestätigungen für eine schnelle Suche vorgeneriert haben. Wir haben diese Liganden gegen den 14-Merkmal-Pharmakophor gescreent, mit der Voraussetzung, dass mindestens ein Merkmal aus jeder der beiden erforderlichen Gruppen sowie mindestens zwei Merkmale aus den verbleibenden acht Merkmalen übereinstimmen müssen, um einen Treffer zu identifizieren. Dies soll eine vielfältige Erforschung unterschiedlicher Bindungsmodi der Liganden ermöglichen und gleichzeitig sicherstellen, dass nur Posen beibehalten werden, die mindestens zwei Wechselwirkungen mit signifikanten Resten eingehen können, die in der Alanin-Scanning-Mutagenese identifiziert wurden. Der Algorithmus untersucht im Wesentlichen über 12.000 mögliche Pharmakophor-Kombinationen. Wir haben dann die am besten passenden Treffer für jede Pharmakophorkombination innerhalb des Proteins minimiert. Schließlich haben wir die Bindungsenergien der optimierten, von Pharmakophoren abgeleiteten Posen mit CHARMm mit einem impliziten Lösungsmittelmodell von Generalized Born mit Molecular Volume (GBMV) bestimmt. Diese Methode ist effektiv eine molekulare Docking-Berechnung (wie in unserem vorherigen Blog), aber hier haben wir unser optimiertes Pharmakophor-Modell verwendet, um die angedockten Ergebnisse einzuschränken und zu filtern.
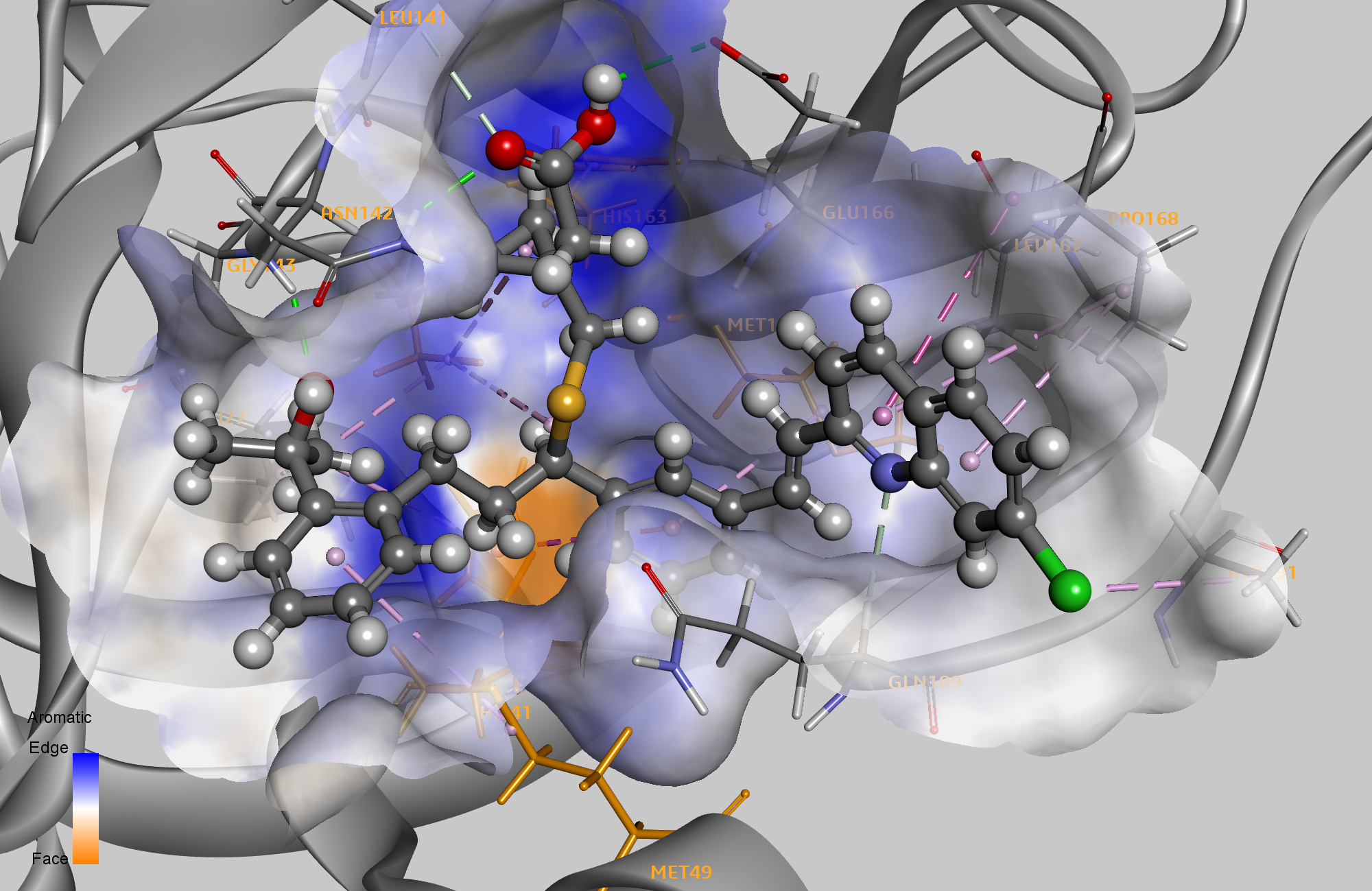
Wir haben die nicht-bindenden Wechselwirkungen jeder Pose berechnet und gefiltert, um uns nur auf die Posen mit Interaktionen mit vier Schlüsselresten zu konzentrieren – HIS41, MET49, CYS145 und MET165. HIS41, MET49 und MET165 waren die drei Reste, die mindestens vier zuvor identifizierten Komplexen gemeinsam waren, und CYS145 ist der zweite Rest in der signifikanten HIS41/CYS145-katalytischen Dyade, die in jeder Untereinheit des Hauptprotease-Homodimers gefunden wird. Wir sortierten die Treffer mit der berechneten freien Bindungsenergie, um die zehn plausibelsten Kandidaten für die weitere Untersuchung zu identifizieren. Ritonavir war der einzige gemeinsame Ligand, der hier und im vorherigen Blog identifiziert wurde. Ritonavir hatte die siebtbeste freie Bindungsenergie und war zuvor auch in der Docking-Studie auf Platz sieben gelandet. Ritonavir durchläuft derzeit eine Reihe von klinischen Studien zu COVID-19. 3
Video 1 :Am besten bewertete Pose für Ritonavir, das mit HIS41, MET49, SER144, CYS145, MET165, GLU166 und GLN189 interagiert.
Der Ligand mit der besten Bewertung in der hier vorgestellten Methode war Montelukast, ein Cysteinylleukotrien-Rezeptor-Antagonist. In einer kürzlich veröffentlichten Veröffentlichung wurde die Hypothese aufgestellt, dass es verwendet werden kann, um das Fortschreiten der Krankheit bei einer COVID-19-Infektion zu begrenzen. 4
Drei weitere Wirkstoffe mit der höchsten Punktzahl waren die Medikamente Telmisartan, Moexipril und Hydroxycloroquin. Das Potenzial all dieser Verbindungen als wiederverwendete Medikamente für COVID-19 wurde kürzlich auf den Prüfstand gestellt, 5 wobei Hydroxychloroquin die meisten Kontroversen auslöste. Ein weiterer Unterschied zwischen den Top-Ten-Hitlisten der beiden Methoden ist die Vielfalt der enthaltenen Wirkstoff-Targets. Zu den Top-Ten-Docking-Ergebnissen gehören sieben HCV- oder HIV-Protease-Inhibitoren. Die nach Pharmakophoren priorisierte Hitliste umfasst Medikamente mit sieben verschiedenen Zielklassen.
Unser von Pharmakophoren abgeleitetes virtuelles Screening hat eine priorisierte Hitliste erstellt, die mehrere neue potenzielle COVID-19-Medikamente enthält, die in unseren früheren Docking-Arbeiten nicht identifiziert wurden. Mit diesem Ansatz demonstrieren wir die Nutzung von in silico Alanin-Scanning-Mutagenese als nützliche Technik zur Verfeinerung des Pharmakophormodells. Wir haben auch die Anforderung auferlegt, dass während des Screening-Prozesses bestimmte Merkmale für eine Passform vorhanden sein müssen. Die Ergebnisse dieser beiden Studien sind nicht direkt vergleichbar – nicht so sehr aufgrund der Unterschiede in den grundlegenden Algorithmen, sondern eher aufgrund der Implementierung unterschiedlicher unterstützender Strategien.
Der kurze Vergleich der Ergebnisse zwischen den beiden Methoden zeigte wenig Überschneidungen bei den Ergebnissen, auf die man sich konzentrieren sollte. Man könnte sagen, dass die Gemeinsamkeit von Ritonavir, die durch beide Methoden identifiziert wurde, Beweise für die Auswahl dieses Kandidaten für weitere Studien liefert. Frühere Studien haben gezeigt, dass die Verwendung eines Konsensus von Protein-Liganden-Docking-Scoring-Funktionen die Identifizierung mutmaßlicher Wirkstoffkandidaten verbessert. 6 Aus diesem Grund könnten Forscher die einzigartigen Verbindungen mit der besten Punktzahl jeder Methode verwenden, um Verbindungen für experimentelle Tests zu priorisieren.
Zusammenfassend lässt sich sagen, dass das von Pharmakophoren abgeleitete virtuelle Screening eine ergänzende und ergänzende Methode zum Andocken bietet, die zu einem Konsens und einem größeren Vertrauen in die Kandidatenauswahl für die experimentelle Validierung beiträgt. Abgesehen von der Dringlichkeit, einen Wirkstoffkandidaten zur Wiederverwendung bereitzustellen, kann diese pharmakophorgesteuerte Methode auch Liganden identifizieren, die für die nachfolgende Leitstrukturoptimierung vielfältiger sind.
Biologie
- Nikotinpflaster
- Virtuelle Bestandsaufnahme und 3D-Druck:das Bedürfnis nach Sicherheit
- C# for-Schleife
- C für Schleife
- Nanofasern und Filamente für eine verbesserte Wirkstoffabgabe
- Generatives Therapeutikum-Design
- Wiederverwendung von Medikamenten
- Universal Robots startet die „weltgrößte“ virtuelle Konferenz für kollaborative Roboter
- Leiterplattenfertigung für 5G
- 3D-gedruckte Mikroroboter versprechen Arzneimittelabgabe