


Wiederverwendung von Medikamenten
Hemmung der SARS-CoV-2-Hauptprotease
Die Entwicklung eines neuen Medikaments ist ein kostspieliger und langwieriger Prozess. Da die Welt heute mit einer Pandemie konfrontiert ist, besteht die dringende Notwendigkeit, schnell Medikamente zu identifizieren, die die Virusverbreitung stoppen. Die Wiederverwendung von Medikamenten bietet eine attraktive Alternative zu diesem langwierigen Prozess, indem versucht wird, herauszufinden, ob ein Medikament, von dem bekannt ist, dass es beim Menschen sicher ist, zur Behandlung neuer Krankheiten verwendet werden könnte.
Während die alleinige Verwendung solcher neu positionierter Medikamente möglicherweise keinen signifikanten klinischen Nutzen bringt, könnte eine sorgfältige Kombination von Medikamenten, die auf mehrere Proteine abzielen, die für die Virusreplikation und -proliferation entscheidend sind, sehr effektiv sein, wie es bei HIV in den 1990er Jahren der Fall war. Die dringende Frage ist, welche Kombination effektiver wäre?
Hier versuchen wir, die Struktur des aktiven Zentrums der SARS-CoV-2-Protease zu verstehen, indem wir sie mit bestehenden Strukturen der SARS-CoV-Protease, die mit mikromolaren Inhibitoren komplexiert ist, vergleichen, damit wir die Schlüsselinteraktionen besser verstehen können, die für die Herstellung eines guten Inhibitors erforderlich sind für SARS-CoV-2-Protease.
Anschließend führten wir ein virtuelles Screening-Experiment mit einer Bibliothek von FDA-zugelassenen Medikamenten durch, um zu sehen, ob einige davon voraussichtlich an die Protease binden. Wir haben untersucht, wie sie voraussichtlich an die SARS-CoV-2-Protease binden und somit in einer Kombinationstherapie verwendet werden könnten.
SARS-CoV-2 Proteine
Das SARS-CoV-2-Genom von kranken Patienten wurde schnell isoliert und sequenziert, um die Sequenzen möglicher Proteinziele bereitzustellen. Diese Proteine weisen eine hohe Sequenzähnlichkeit mit SARS-CoV-Proteinen auf und zunächst begannen Forschungsgruppen mit dem Aufbau von Homologiemodellen. Jetzt sehen wir, dass immer mehr dieser experimentell abgeleiteten (Röntgen- und Kryo-EM) Strukturen verfügbar werden.
Eines der am besten charakterisierten Wirkstoffziele unter den Coronaviren ist die Hauptprotease:Mpro, auch 3CL-Protease genannt. 1 Zusammen mit den Papain-ähnlichen Proteasen ist dieses Enzym essentiell für die Verarbeitung der Polyproteine, die von der viralen RNA translatiert werden. 2 Es spaltet das Aminosäurerückgrat an 11 Schnittstellen des großen Polyproteins 1ab (Abb. 1).
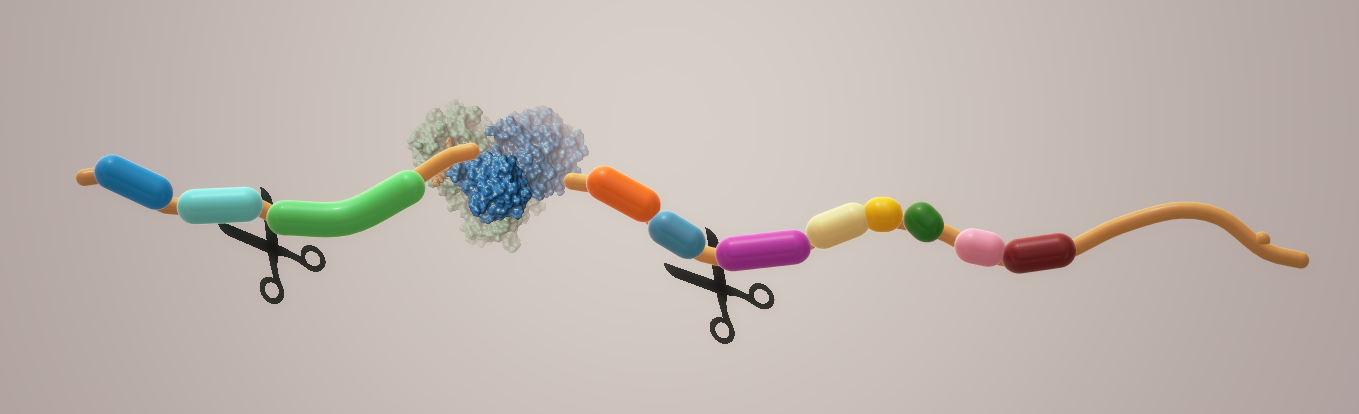
Eine Hemmung der Aktivität dieses Enzyms würde die Virusreplikation blockieren. Da keine humanen Proteasen mit einer ähnlichen Spaltungsspezifität bekannt sind, ist die Wahrscheinlichkeit, dass Inhibitoren dieses Targets toxisch sind und Nebenwirkungen verursachen, viel geringer.
Strukturbasiertes Medikamentendesign für die Wiederverwendung von Medikamenten:Hauptprotease
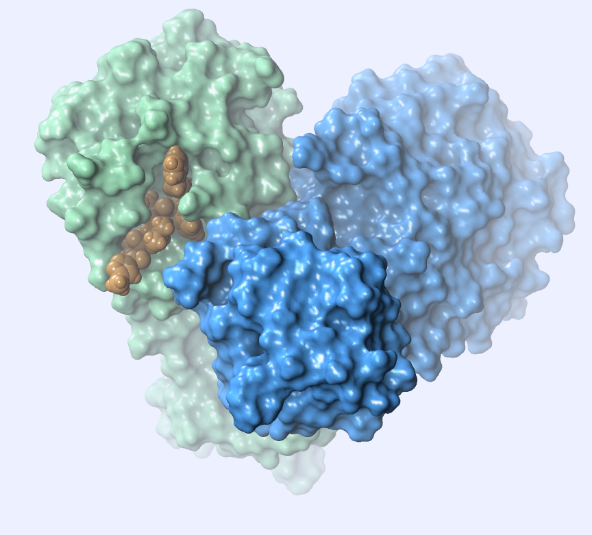
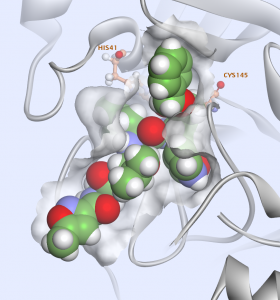
Diese Hauptprotease ist ein Homodimer (Abb. 2) und jede Untereinheit enthält eine katalytische His41/Cys145-Dyade.
SARS-CoV-2 teilt 96,1 % Identität und 99 % Ähnlichkeit mit der SARS-CoV-Protease. (Abb. 3) Es gibt nur einen Aminosäureunterschied im aktiven Zentrum:Rest 46 in SARS-CoV-2 ist ein Serin anstelle eines Alanins in SARS-CoV.
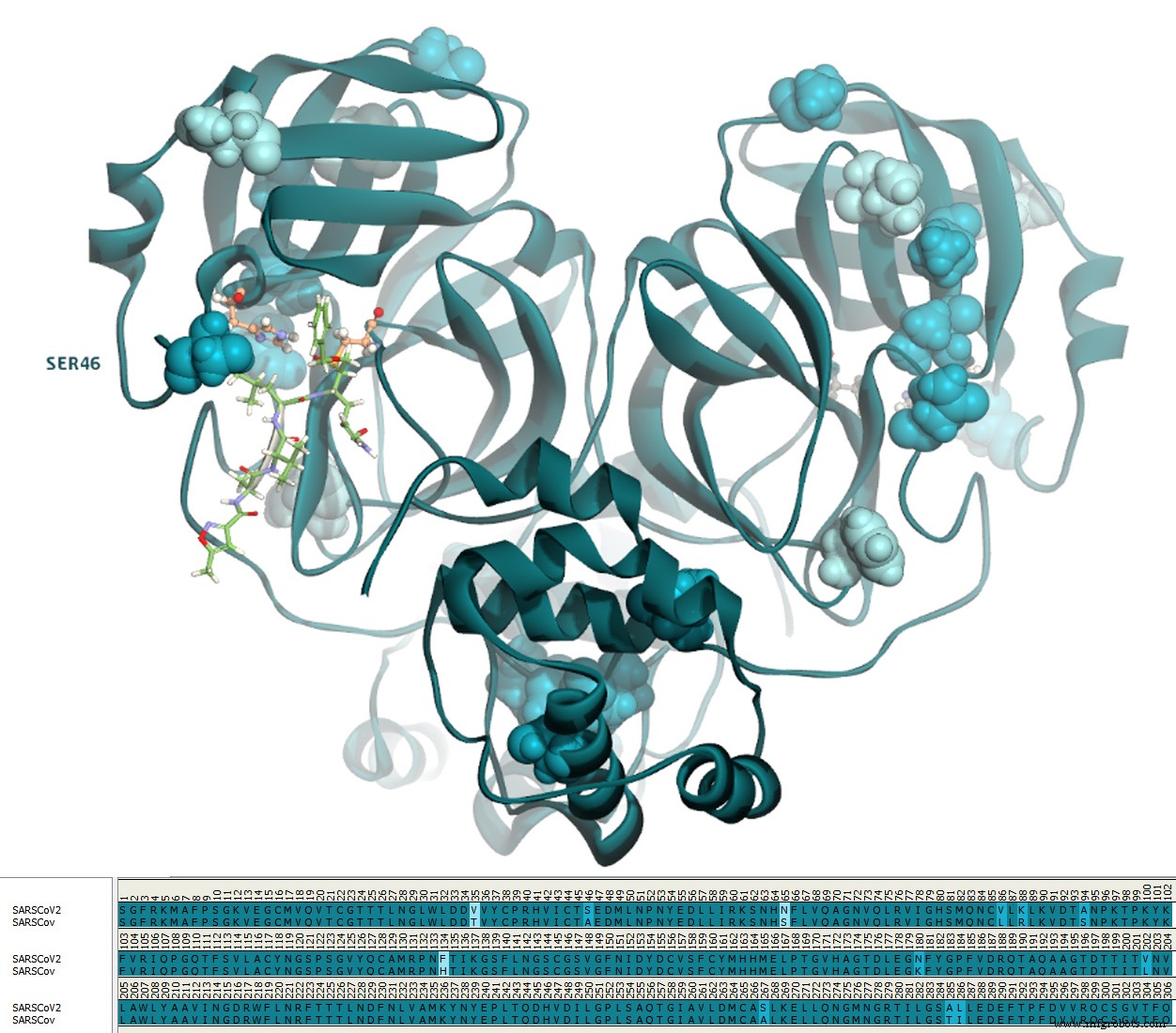
Es gibt viele Strukturen von SARS-CoV Mpro, die mit Liganden komplexiert sind, die an der PDB verfügbar sind, und die Liganden mit einer mikromolaren Bindungsaffinität sind alle kovalente Liganden.
Die aktiven Zentren der Hauptprotease sind bei Coronaviren stark konserviert und bestehen normalerweise aus vier Zentren (S1′, S1, S2 und S4) 3 (Abb. 4).
Im Fall der PDB-Strukturen 2ZU4 und 2GX4, für die Liganden Ki-Werte von 0.038 μM bzw. 0.053 μM aufweisen, kontaktiert das Thiol von Cystein 145 in der S1′-Stelle Inhibitoren mit einer kovalenten Bindung. Im Vergleich zu anderen Inhibitoren mit geringerer Affinität scheint dies für eine höhere Affinität wichtig zu sein.
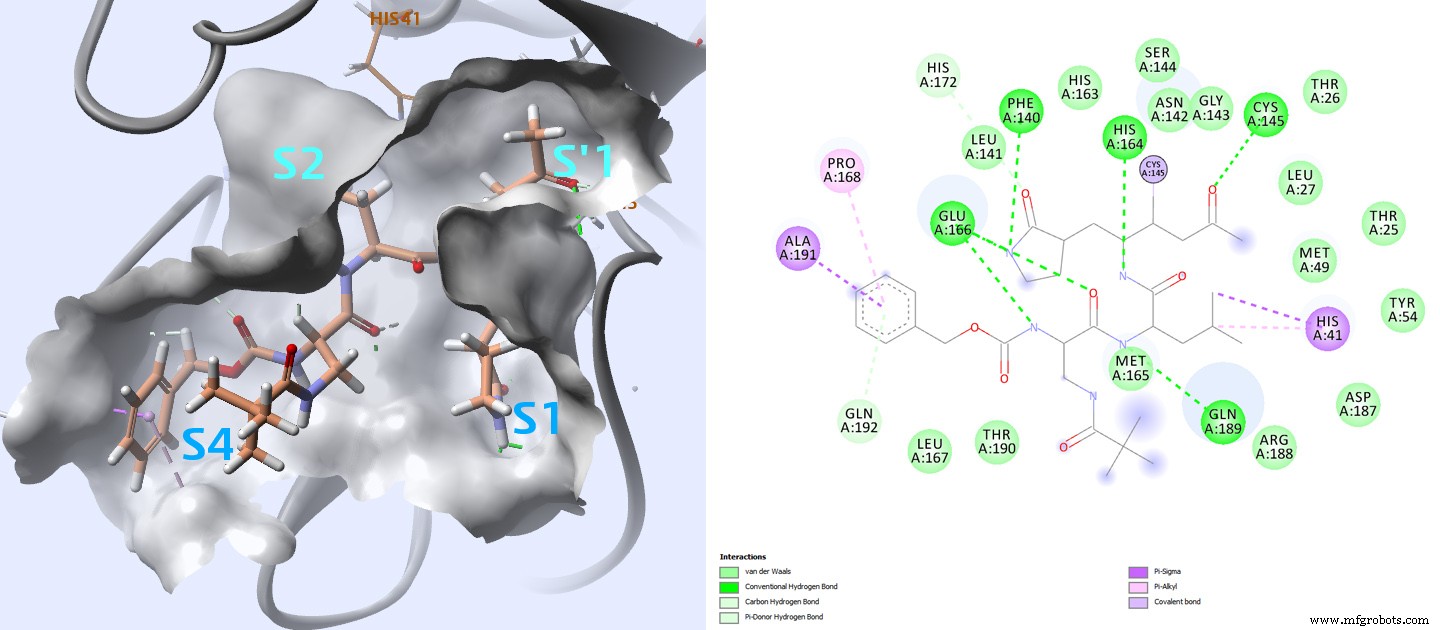
Eine sorgfältige Untersuchung, wie all diese Liganden mit dieser Protease interagieren, kann Informationen über Schlüsselinteraktionen liefern, die bei der Analyse der Docking-Ergebnisse überwacht werden müssen.
Virtuelles Screening
Ausnutzung einer hochauflösenden Kristallstruktur des SARS-COV-2-Protease-Dimers im Komplex mit einem kovalent gebundenen peptidähnlichen Inhibitor N3, der im Februar freigesetzt wurde (6LU7) 4 , haben wir diese Struktur verwendet, um ein virtuelles Screening-Experiment durchzuführen. Der cokristallisierte Inhibitor N3 bindet in einer erweiterten Konformation in der Substratbindungstasche. (Abb. 5)
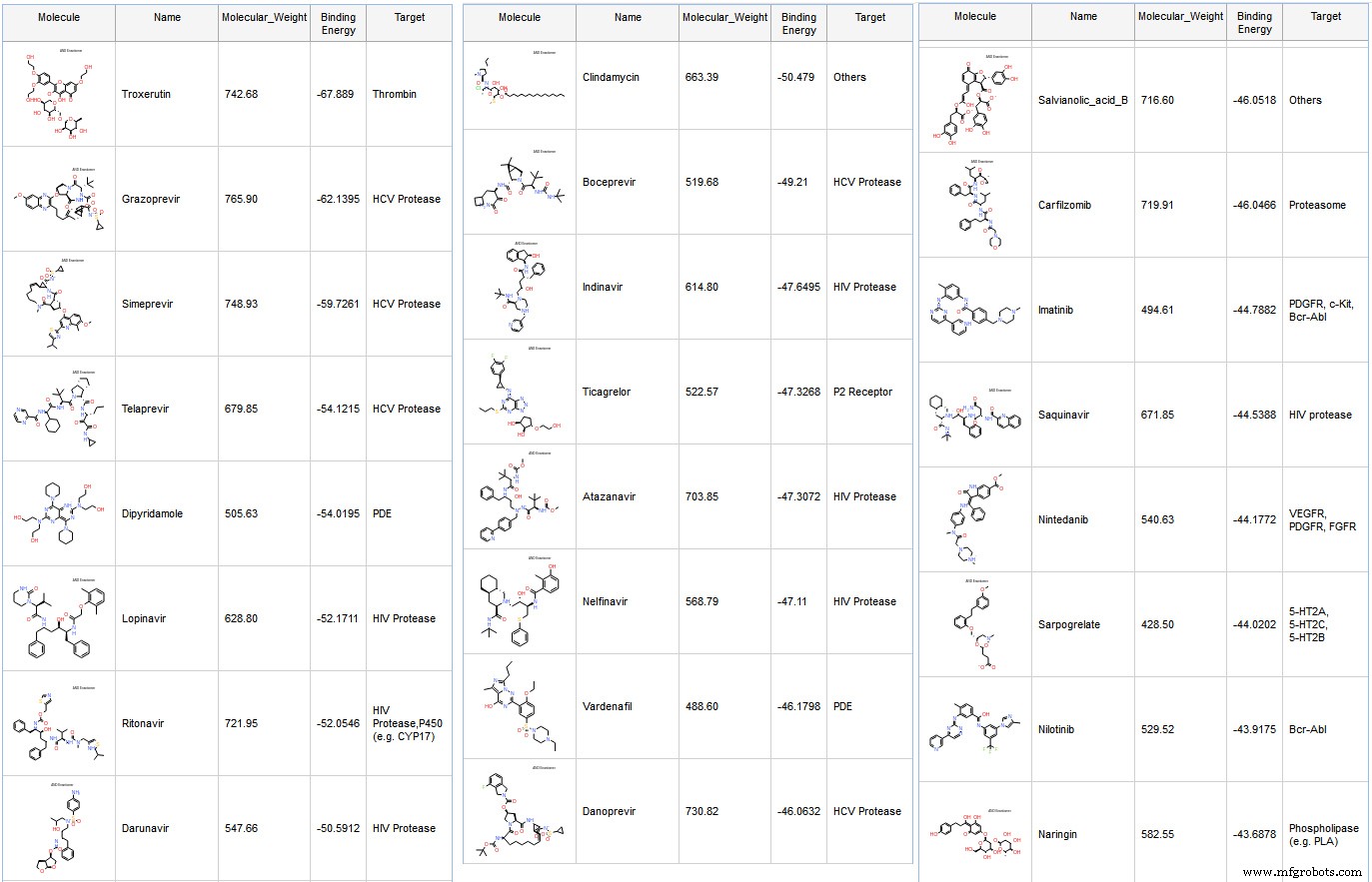
Wir begannen mit einer Bibliothek mit von der FDA zugelassenen Medikamenten mit 2.684 Verbindungen. Wir behielten Verbindungen mit einem Molekulargewicht von weniger als 800 kDa bei und führten das Andocken mit GOLD vom CCDC durch. Später berechneten wir Bindungsfreie Energien mit CHARMM und GBMV implizitem Lösungsmittel. Für jede Pose führten wir zuerst eine In-Situ-Ligandenminimierung mit einer Kugel von 14 Å Durchmesser für die Restflexibilität durch und schätzten die Ligandenentropie bei der Berechnung der freien Bindungsenergie. Die meisten der angedockten Verbindungen sind bekannte HCV- und HIV-Protease-Medikamente. (Abb. 6)
Viele Verbindungen stellen Kontakte in den 4 Subsites her:S1, S’1, S2, S4 und einige mit HIS 41 und CYS 145, insbesondere die HIV-Protease-Inhibitoren.
Die am besten bewertete Verbindung ist Troxerutin, ein Flavonoid.
- Flavonoide hemmen nachweislich einige Proteasen 5 und vor kurzem IC50 Werte wurden aus den dosisabhängigen Hemmkurven von Herbacetin, Rhoifolin und Pectolinarin auf SARS-CoV berechnet. Die gemessenen Werte waren 33,17, 27,45 bzw. 37,78 µM.
Dipyridamol (Abb. 7), die an fünfter Stelle eingestufte Verbindung, wurde kürzlich in einem Vorabdruck erwähnt>in vitro.
Abbildung 7 :3D-Rendering und 2D-Interaktionskarte von Dipyridamol in der Hauptprotease. Wechselwirkungen mit katalytischen Rückständen sind vorhanden und mit einigen der zuvor für SARS-CoV-Inhibitoren erwähnten Rückstände.
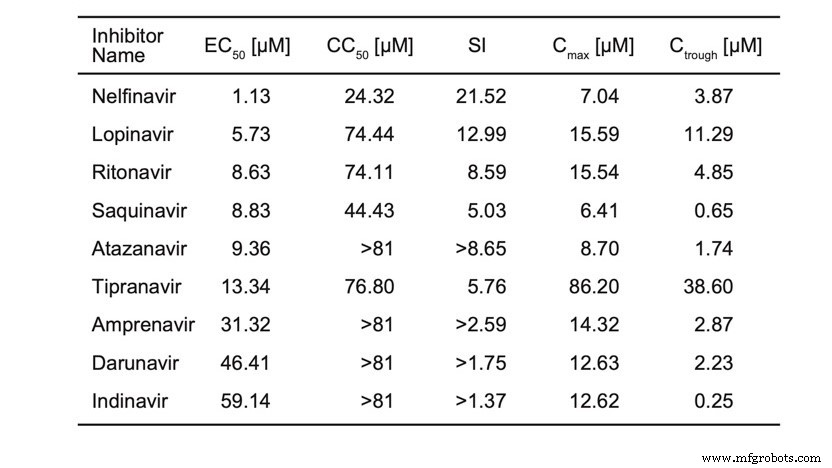
Ein kürzlich veröffentlichter Preprint berichtete über zelluläre Assays mehrerer HIV-Protease-Inhibitoren:„Nelfinavir hemmt die Replikation des schweren akuten respiratorischen Syndroms Coronavirus 2 in vitro .”
Wir berichten die Tabelle aus dem Preprint (Tabelle 1). Dies sind nicht IC50, sondern EC50, daher sagt es nicht aus, wie gut diese Moleküle an die Protease binden. Dies zeigt jedoch, dass all diese Verbindungen die Replikation von SARS-CoV-2 hemmen und einige aktiver sind als andere.
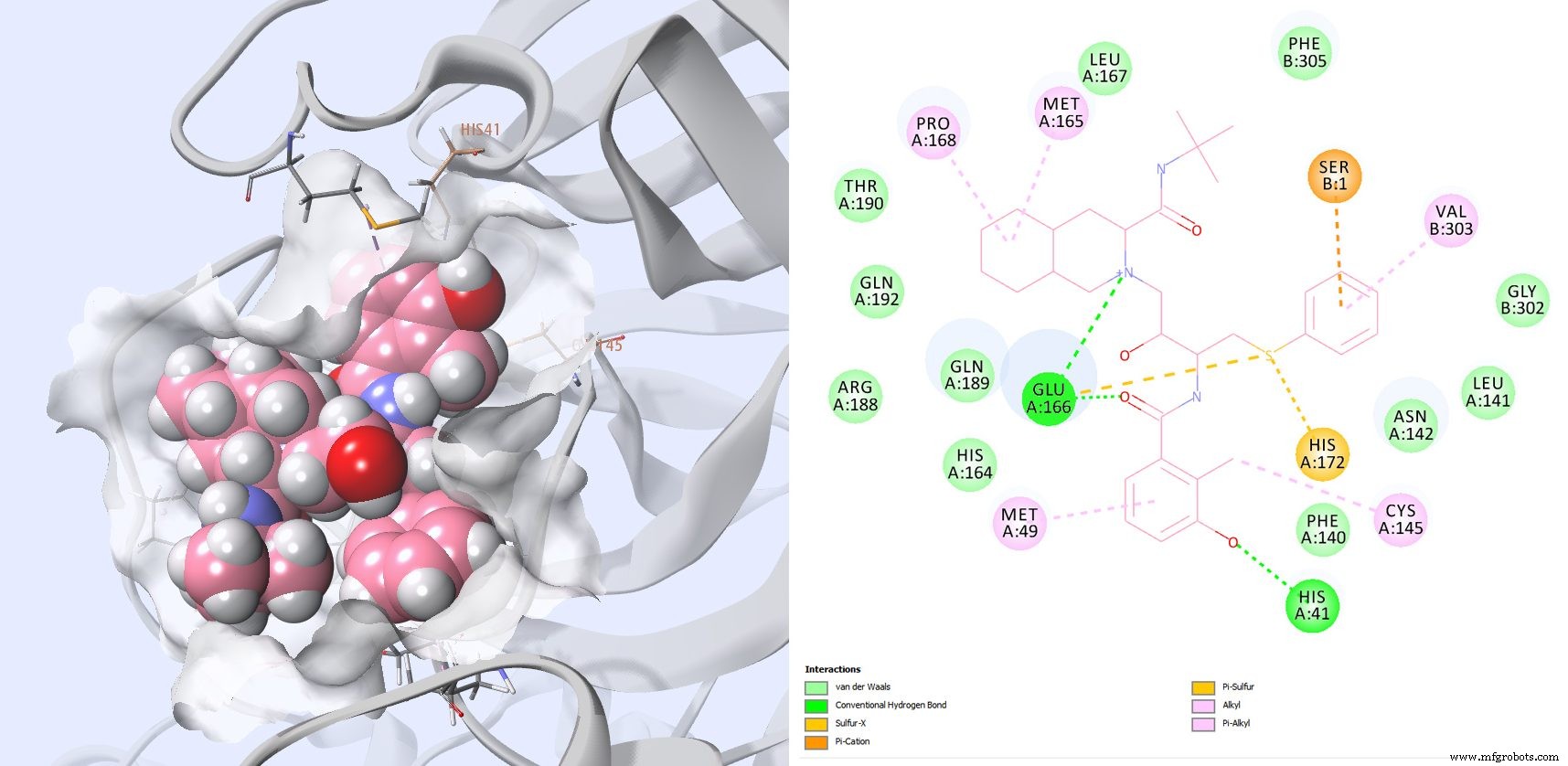
Ritonavir und Lopinavir wechselwirken mit vielen Rückständen, die zuvor in anderen Verbindungen beobachtet wurden, die auf die SARS-CoV-Protease abzielen (Abb. 8 und 9). Obwohl mehrere klinische Studien mit einer Kombination dieser beiden Moleküle laufen, sind leider keine IC50-Daten für Ritonavir oder Lopinavir zu SARS-CoV-2 verfügbar.
Abbildung 8 :Am besten bewertete Pose für Ritonavir:und 2D-Interaktionsdiagramm, das zeigt, dass Ritonavir mit den beiden katalytischen Resten interagiert:CYS145 und HIS41, aber auch GLU166, PRO168 und GLN189, wie bei anderen bekannten Inhibitoren von SARS-CoV.
Abbildung 9 :Am besten bewertete Pose für Lopinavir:und 2D-Interaktionsdiagramm, das zeigt, dass Lopinavir mit den beiden katalytischen Resten interagiert:CYS145 und HIS41, aber auch GLU166, PRO168 und GLN189, wie bei anderen bekannten Inhibitoren von SARS-CoV.
Nelfinavir (Abb. 10) hat in unseren Berechnungen eine hohe vorhergesagte Bindungsaffinität und wurde in einem aktuellen Preprint auch gegen COVID19 charakterisiert:„Nelfinavir ist aktiv gegen SARS-CoV-2 in Vero E6-Zellen.“
Ein zweiter Vorabdruck zeigt, dass Atazanavir auf SARS-CoV-2-infizierten Zellen aktiv ist:„Atazanavir hemmt die SARS-CoV-2-Replikation und die entzündungsfördernde Zytokinproduktion.“
Nachdem wir mit dieser Arbeit begonnen hatten, wurde eine PDB-Struktur 6W63 veröffentlicht, die einen neuen Inhibitor für SARS-CoV-2 enthält (Abb. 11). Es gibt keine veröffentlichten Informationen über die Bindungsaffinität dieser Verbindung zum SARS-CoV-2 Mpro. Die Seitenketten in der Bindungsstelle haben fast die gleiche Orientierung wie bei 6LU7. Wir haben unsere Verbindungen angedockt und sie wie zuvor mit MMGBSA-Ansätzen neu bewertet und sehr ähnliche Ergebnisse gefunden.
Abbildung 11 :PDB-Struktur 6W63:SARS-CoV-2-Hauptprotease im Komplex mit Inhibitor X77.
Perspektiven
Die Bindungsstelle der SARS-CoV-2-Protease ist sehr groß, enthält vier Unterstellen und kann viele verschiedene Liganden mit einer mäßigen Bindungsaffinität aufnehmen. Wie hier erläutert, ist das virtuelle Screening ein nützliches Werkzeug, um mögliche Inhibitoren zu identifizieren, und konnte uns eine strukturelle Erklärung dafür liefern, wie mögliche Inhibitoren mit der Protease interagieren könnten.
All diese Hypothesen erfordern jedoch eine Bestätigung durch experimentelle Beweise, wie die Messung von IC50, um robustere Modelle zu erstellen.
Hier haben wir kein kovalentes Andocken durchgeführt, aber da die aktivsten Verbindungen gegen die SARS-CoV-Protease kovalent waren, könnte dies eine Voraussetzung für starke Inhibitoren sein. Kovalente Inhibitoren weisen im Vergleich zu reversiblen Inhibitoren auch Vorteile auf, wie z. B. eine starke Zielaffinität und eine verlängerte Wirkdauer bei Patienten.
Damit ein Medikament bei einem Patienten wirksam ist, müssen wir andere pharmakologische Aspekte wie die Pharmakokinetik berücksichtigen. Es wird notwendig sein, die Fähigkeit dieser Medikamente zu bewerten, die angestrebten Plasma- und Lungenkonzentrationen nach einer zugelassenen Dosierung beim Menschen zu erreichen.
Es lag nicht im Rahmen dieser Arbeit, andere strukturbasierte Designforschungen zu kommentieren, da dies für die aktuelle Epidemie nicht zeitgemäß wäre. Unser Ziel war es, leicht anwendbare Therapeutika für potenzielle Medikamentenkandidaten gegen COVID19 zu bewerten.
Wir danken dem Cambridge Crystallographic Data Centre (CCDC) für die Erlaubnis, GOLD für das virtuelle Screening in dieser Arbeit zu verwenden. GOLD hat sich beim virtuellen Screening, der Leitstrukturoptimierung und der Identifizierung des richtigen Bindungsmodus aktiver Moleküle als erfolgreich erwiesen. Eine Schnittstelle zu GOLD ist in Discovery Studio verfügbar.
Biologie
- China will Patentverknüpfung etablieren
- Die US-amerikanische Arzneimittelversorgungskette in der Krise:Lösungen für Engpässe
- COVID-19 hat fatale Mängel in der US-amerikanischen Arzneimittelversorgungskette aufgedeckt
- Wie Blockchain im Kampf gegen gefälschte Medikamente helfen könnte
- Anwenden von Blockchain und maschinellem Lernen auf Arzneimittellieferketten
- All-in-One-Test für die COVID-19-Überwachung
- Neuer photonischer Effekt könnte Arzneimittelentwicklung beschleunigen
- Smartwatch verfolgt Medikamentenspiegel
- 5 Ws des SARS-CoV-2 RapidPlex Sensors
- 3D-gedruckte Mikroroboter versprechen Arzneimittelabgabe