


Metallanalyse durch optische Emissionsspektroskopie
Metallanalyse durch optische Emissionsspektroskopie
Optische emissionsspektroskopische Techniken haben ihren Ursprung in Experimenten, die Mitte des 19. Jahrhunderts durchgeführt wurden, aber sie gehören nach wie vor zu den nützlichsten und flexibelsten Mitteln zur Durchführung der Elementaranalyse. Freie Atome emittieren Licht in einer Reihe enger Wellenlängenintervalle, wenn sie in eine energetische Umgebung gebracht werden. Diese als Emissionslinien bezeichneten Intervalle bilden ein Muster, das als Emissionsspektrum bekannt ist und das Merkmal des Atoms ist, das es erzeugt. Die Intensitäten der Linien sind normalerweise proportional zur Anzahl der sie erzeugenden Atome. Das Vorhandensein eines Elements in einer Probe wird durch das Vorhandensein einer oder mehrerer seiner charakteristischen Linien im Licht von der Anregungsquelle angezeigt. Die Konzentration dieses Elements kann durch Messung der Linienintensität bestimmt werden. Somit bildet das charakteristische Emissionsspektrum die Grundlage für die qualitative Elementanalyse und die Messung der Intensitäten der Emissionslinien die Grundlage für die quantitative Elementanalyse. Die Emissionsspektren für Eisen und Lithium sind in Abb. 1 dargestellt.

Abb. 1 Emissionsspektrum für Eisen und Lithium
Optisches Emissionsspektrometer, auch als optisches Emissionsspektrometer bekannt, wird normalerweise für (i) die quantitative Bestimmung von Haupt- und Spurenelementbestandteilen in verschiedenen Probentypen und (ii) die qualitative Elementaranalyse verwendet. Anwendungsbeispiele umfassen (i) schnelle Bestimmung von Konzentrationen von Legierungselementen in Stählen und anderen Legierungen, (ii) Elementaranalyse von geologischen Materialien, (iii) Bestimmung von Spurenverunreinigungskonzentrationen in Halbleitermaterialien, (iv) Verschleißmetallanalyse in Ölen, (v) Bestimmung von Alkali- und Erdalkalikonzentrationen in wässrigen Proben und (vi) Bestimmung von Calcium in Zement.
Proben liegen in Form von leitfähigen Feststoffen (Lichtbögen, Funken und Glimmentladungen), Pulvern (Lichtbögen) und Lösungen (Flammen) vor. Die Probengröße hängt von der spezifischen Technik ab und variiert von etwa 0,000001 Gramm bis zu mehreren Gramm. Die Probenvorbereitung erfolgt durch Bearbeiten oder Mahlen (Metalle), Auflösen (für Flammen) und Aufschluss oder Veraschen (organische Proben).
Einschränkungen der optischen emissionsspektroskopischen Techniken sind (i) einige Elemente sind schwierig oder unmöglich zu bestimmen, wie Stickstoff, Sauerstoff, Wasserstoff, Halogene und Edelgase, (ii) die Probenform muss mit einer bestimmten Technik kompatibel sein, und (iii ) liefern alle Methoden matrixabhängige Antworten. Die geschätzte Analysezeit reicht von 30 Sekunden bis zu mehreren Stunden, je nach Anforderungen an die Probenvorbereitung.
Zu den Fähigkeiten verwandter Techniken gehören (i) Röntgenfluoreszenz ist für die Elementaranalyse von Massen- und Nebenbestandteilen und erfordert eine ausgeklügelte Datenreduktion für die quantitative Analyse und ist für leichte Elemente (Ordnungszahl 9) nicht nützlich, (ii) induktiv gekoppeltes Plasma (ICP) Emissionsspektroskopie dient der schnellen quantitativen Elementaranalyse mit Nachweisgrenzen von Teilen pro Milliarde, wobei sich die Proben in Lösung befinden müssen, und ist für Wasserstoff, Stickstoff, Sauerstoff, Halogenide und Edelgase nicht geeignet, (iii) die Gleichstrom-Plasma-Emissionsspektroskopie ist ähnlich in der Leistung gegenüber der ICP-Emissionsspektroskopie, und (iv) die Atomabsorptionsspektroskopie ist eine Einkanaltechnik, die für die Multielementanalyse ineffizient ist, aber für die meisten Elemente eine günstige Empfindlichkeit und Präzision aufweist.
Im weiteren Sinne umfasst die optische Emissionsspektroskopie die optische ICP-Emissionsspektroskopie, die einen ICP als Anregungsquelle verwendet. Die Begriffe „optische Emissionsspektroskopie“ und „optische Emissionsspektroskopie“ beziehen sich jedoch normalerweise auf optische Emissionsspektroskopie unter Verwendung von Funkenentladung, Gleichstrombogenentladung, Glimmentladung oder Flammenquelle zum Erzeugen der Anregungsentladung. In diesem Artikel wird die optische Spektroskopie mit Funkenentladung diskutiert, da sie in der Stahlindustrie eingesetzt wird.
Viele optische Emissionsspektroskope verfügen über eine „Impulsverteilungsanalyse“ (PDA), um die Reproduzierbarkeit (Genauigkeit) der Messung zu verbessern. Dieses Verfahren beinhaltet eine statistische Verarbeitung der durch Funkenimpulse erzeugten Emissionsspektren, die aus Funkenentladungen in einer Argonatmosphäre erhalten werden. Das optische Emissionsspektroskop bietet eine schnelle Elementaranalyse von festen Metallproben und ist damit unverzichtbar für die Qualitätskontrolle in der Stahlherstellung
Allgemeine Grundsätze
Das charakteristische Spektrum, das ein Atom erzeugt, spiegelt die elektronische Struktur des Atoms wider. Änderungen in der Energie der Valenz- oder Außenschalenelektronen führen zu den in der Emissionsspektroskopie verwendeten Atomlinien. Jedes Atom hat einen Grundzustand, in dem alle seine Elektronen Positionen mit minimaler potentieller Energie einnehmen. Wenn ein Atom Energie absorbiert, können ein oder mehrere der äußeren Elektronen auf höhere Energien gebracht werden, wodurch ein angeregter Zustand erzeugt wird. Die Energie eines Atomzustands ist eine Funktion der Energien der einzelnen Elektronen und von Energieänderungen, die aus Wechselwirkungen zwischen den Elektronen resultieren. Jede mögliche Kombination von Elektronenkonfigurationen erzeugt einen spektroskopischen Term, der den Zustand des Atoms beschreibt. Abb. 2 zeigt das Prinzip des Emissionsspektrums am Beispiel eines Lithiumatoms.
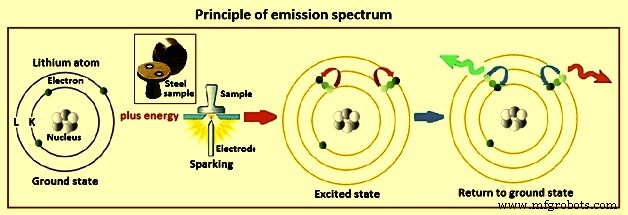
Abb. 2 Prinzip des Emissionsspektrums
Elektronenenergieniveaus – Die einfachsten Atome wie Wasserstoff und die Alkalimetalle haben nur ein Elektron außerhalb einer gefüllten Schale. Die einfachen Elektronenkonfigurationen dieser Atome erzeugen mehrere mögliche Terme. Atomemissionslinien entstehen, wenn das Atom einen spontanen Übergang von einem angeregten Zustand in einen anderen niedrigeren Energiezustand erfährt. Nicht alle möglichen Kombinationen von Zuständen erzeugen Emissionslinien. Nur Übergänge, die quantenmechanisch abgeleiteten Auswahlregeln gehorchen, treten spontan auf. Verschiedene Faktoren steuern die relativen Intensitäten der Linien. Diese Übergänge zwischen einem niedrig angeregten Zustand und dem Grundzustand, die als Resonanzübergänge bezeichnet werden, ergeben im Allgemeinen die intensivste Emission.
Die Energie des angeregten Elektrons nimmt mit abnehmendem Abstand zwischen den angeregten Zuständen zu, bis es eine Ionisationsgrenze erreicht. An diesem Punkt ist das Elektron nicht mehr an das Atom gebunden und kann einen kontinuierlichen Energiebereich annehmen. Solche ungebundenen Elektronen können in gebundene Zustände übergehen. Da der obere Zustand des Übergangs nicht auf diskrete Werte beschränkt ist, wird das Licht solcher Übergänge kontinuierlich über einen Bereich von Wellenlängen gestreut.
Die Ionisationsgrenze für das Atom entspricht dem Grundzustand des einfach geladenen Ions. Die Anregung der verbleibenden gebundenen Elektronen ergibt ein neues Termsystem und einen neuen Liniensatz. Ionisierung und Anregung können fortgesetzt werden, bis ein Atom vollständig von seinen Elektronen befreit ist. In praktischen Emissionsquellen geht die Ionisation selten über die Entfernung von zwei Elektronen hinaus, und in den meisten Fällen braucht nur die erste Stufe der Ionisation betrachtet zu werden. Anstelle einer neutralen Atomlinie wird jedoch normalerweise eine Linie aus dem ersten Ionenspektrum in der Analyse verwendet.
Spektrale Überlappung – Die Verwendung der Atomemission für die Elementaranalyse erfordert die Messbarkeit der Emissionsintensität einer interessierenden Linie, unabhängig von überlappenden Emissionen anderer Spezies in der Probe. Die Wahrscheinlichkeit einer unerwünschten Überlappung hängt von der Anzahl der Linien im Spektrum und von der Wellenlängenspreizung oder Linienbreite jedes Übergangs ab. Wenn alle atomaren Termsysteme so einfach sind wie das für Lithium in Abb. 2 gezeigte, ist die Wahrscheinlichkeit einer spektralen Überlappung gering. Allerdings ist Lithium eines der einfachsten Atome.
Atome mit komplexeren elektronischen Strukturen erzeugen entsprechend komplexe Emissionsspektren. Das Eisenspektrum (in Abb. 1 gezeigt) veranschaulicht eine solche spektrale Komplexität. Das Spektrum einer Ionisationsstufe eines einzelnen Elements kann bei ausreichender Anregungsenergie aus Hunderten von Emissionslinien bestehen. Die Komplexität wird noch verstärkt, wenn mehrere Elemente in einer Probe vorhanden sind, die jeweils neutrale und ionische Spektren erzeugen.
Linienverbreiterung – Spektrale Komplexität ist kein Problem, wenn in der Praxis jede Emissionslinie streng monochromatisch ist und Instrumente mit unendlicher spektraler Auflösung verfügbar sind. Die mit einem elektronischen Begriff verbundene Energie ist nicht genau definiert, sondern über einen Wertebereich verteilt. Die Unsicherheit in den Energieniveaus erscheint im Emissionsspektrum als Wellenlängenverbreiterung der Emissionslinien. Mehrere Faktoren bestimmen die Größe der Energiestreuung. Die wichtigsten für die Emissionsspektroskopie sind häufige Kollisionen des emittierenden Atoms oder Ions mit anderen Spezies in der Anregungsquelle und die Platzierung des Emitters in einem inhomogenen elektrischen Feld.
Die erste Art der Linienverbreiterung ist die Kollisionsverbreiterung, während die zweite die Stark-Verbreiterung ist. Ein dritter Typ, die Doppler-Verbreiterung, ergibt sich aus der Bewegung der emittierenden Spezies relativ zu der Vorrichtung, die die Emission erfasst. Bei fester Übergangsenergie hat die Emission, die von einem sich zum Detektor bewegenden Atom aufgezeichnet wird, kürzere Wellenlängen als die, die von einem ruhenden Atom aufgezeichnet wird. Die Emission eines Atoms, das sich vom Detektor wegbewegt, hat längere Wellenlängen. Die relative Größe dieser drei Beiträge zur Linienverbreiterung hängt stark von der Art der Quelle ab, die die Emission anregt. Der Kollisionsbeitrag zur Linienbreite ist hauptsächlich eine Funktion des Quellendrucks. Der Doppler-Beitrag für ein gegebenes Element hängt von der Quellentemperatur ab. Die Größe des Stark-Beitrags hängt von der Dichte der geladenen Spezies in der Nähe des Emitters ab.
Selbstbezogenheit – Atomare Linienprofile, die durch einen der oben genannten Effekte erzeugt werden, können durch Selbstabsorption verändert werden. Bei hohen Atomkonzentrationen in der spektroskopischen Quelle ist die Wahrscheinlichkeit angemessen, dass die von einem Atom emittierte Strahlung von einem anderen gleichartigen Atom absorbiert wird. Die Absorptionswahrscheinlichkeit ist bei Wellenlängen in der Nähe der Mitte des Linienprofils größer als bei Wellenlängen in der Nähe der Flügel. Die unter solchen Bedingungen beobachteten Emissionsprofile sind flacher und breiter als diejenigen, die ohne Selbstabsorption beobachtet werden. Wenn die absorbierenden Atome niedrigere Temperaturen haben als die emittierenden Atome, ist ein Linienprofil ähnlich dem in Abb. 3 gezeigten. Das Doppler-Absorptionsprofil von Tieftemperaturabsorbern ist schmaler als das Emissionsprofil der heißeren Emitter. Dies wird als Selbstumkehrung bezeichnet.
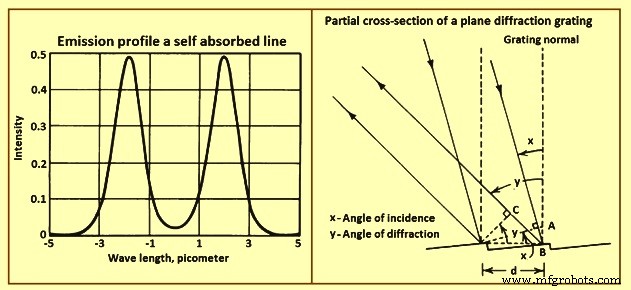
Abb. 3 Emissionsprofil einer selbstabsorbierenden Linie und Teilquerschnitt eines ebenen Beugungsgitters
Molekulare Emission – Das energetisch emittierende Volumen einer spektroskopischen Quelle kann neben freien Atomen auch kleine Moleküle enthalten. Wie die Atome erzeugen die Moleküle eine optische Emission, die die Änderung der Energien der äußeren Elektronen des Moleküls widerspiegelt. Im Gegensatz zu den Atomen haben die Moleküle zahlreiche Schwingungs- und Rotationsniveaus, die mit jedem elektronischen Zustand verbunden sind. Jeder elektronische Übergang im Molekül erzeugt ein Emissionsband, das aus einzelnen Linien besteht, die die Schwingungs- und Rotationsstruktur der am Übergang beteiligten elektronischen Zustände widerspiegeln.
Molekülbanden erscheinen in einem aufgenommenen Spektrum als intensive Kanten, aus denen sich bei höheren oder niedrigeren Wellenlängen weniger intensive Linien mit mit zunehmender Entfernung von der Kante zunehmendem Abstand entwickeln. Der Rand ist der Bandkopf. Molekülbanden bestehen aus vielen eng beieinander liegenden Linien und können einen Bereich des Spektrums dominieren, was die Detektion von Emissionen anderer Arten in diesem Bereich erschwert. Emissionsquellen sind häufig so ausgelegt, dass sie die molekulare Emission minimieren. Seltener werden Bandenintensitäten anstelle von atomaren Linienintensitäten verwendet, um die Konzentration zu messen.
Optische Systeme
Die Atomemission ist nur insofern analytisch nützlich, als die Emission einer Atomart gemessen und ihre Intensität unabhängig von der Emission aus anderen Quellen aufgezeichnet werden kann. Diese Detektion und Quantifizierung erfordert hochauflösende Instrumente zur Wellenlängensortierung. Außerdem muss das Licht, bevor es sortiert werden kann, effizient gesammelt werden, manchmal nur von einer isolierten Region in einer räumlich heterogenen Emissionsquelle.
Instrumente zur Wellenlängensortierung – Das Schlüsselelement moderner Instrumente zur Wellenlängensortierung ist das Beugungsgitter, eine präzise geformte reflektierende Oberfläche mit vielen eng beieinander liegenden parallelen Rillen. Ein teilweiser Querschnitt eines Beugungsgitters ist in Fig. 3 gezeigt. Parallele Lichtstrahlen treffen benachbarte Rillen auf dem Gitter. Die einfallenden Strahlen sind miteinander in Phase. Die vom Gitter gestreuten Strahlen haben verschiedene Wege durchlaufen. Die Differenz der Pfadlängen ist AB + BC.
Bei Winkeln, die eine Wegdifferenz erzeugen, die eine ganze Zahl von Wellenlängen ist, sind die austretenden Strahlen in Phase, und Licht wird in diesem Winkel gebeugt. Bei anderen Winkeln sind die austretenden Strahlen phasenverschoben und destruktive Interferenz tritt auf. Die Winkel, bei denen die Beugung für eine gegebene Wellenlänge stattfindet, können bestimmt werden, indem beachtet wird, dass AB =d sin x und BC =d sin y wobei d der Rillenabstand des Beugungsgitters ist, x der Einfallswinkel ist und y der Beugungswinkel ist. Die Beugungsbedingung ist durch die Gleichung mλ =d.(sin x +/- sin y) gegeben. Das Minuszeichen tritt ein, wenn sich der einfallende und der gebeugte Strahl auf gegenüberliegenden Seiten der Gitternormalen befinden.
Für die Emissionsspektroskopie werden normalerweise zwei Arten von Wellenlängensortiergeräten (Abb. 4) verwendet. Der erste, der Gittermonochromator, wird zur Einkanaldetektion von Strahlung verwendet. Abb. 4 zeigt den Lichtweg durch einen Czerny-Turner-Monochromator, eine typische Konfiguration. Licht tritt durch den Eintrittsspalt in den Monochromator ein und gelangt zum Kollimationsspiegel. Das kollimierte Licht trifft auf das ebene Beugungsgitter und wird unter einem von seiner Wellenlänge abhängigen Winkel gebeugt. Ein Teil des Lichts wird in solchen Winkeln gebeugt, dass es auf den Fokussierspiegel trifft. Es wird dann fokussiert, um eine Reihe von Eintrittsschlitzbildern in der Brennebene des Monochromators zu bilden. Die Position in der Anordnung eines Schlitzbildes hängt von dem Winkel ab, in dem das Licht, das es bildet, aus dem Gitter austritt. Die Wellenlänge des auf dem Austrittsspalt zentrierten Bildes ist durch die Gleichung m gegeben. Lambda =2d.sin q.cos p wobei q der Winkel ist, um den das Gitter gedreht wird, und p der Instrumentenwinkel und der Winkel ist, den eine Linie durch die Mitte des Gitters und die Mitte jedes Spiegels mit der Mittellinie bildet des Instruments. Die Beziehungen zwischen q und p und den Winkeln x und y, die in der ersten Gleichung verwendet werden, sind in Abb. 4 dargestellt. Wenn das Gitter gedreht wird, passieren Bilder verschiedener Wellenlängen nacheinander den Ausgangsschlitz und werden von einer Photovervielfacherröhre detektiert.
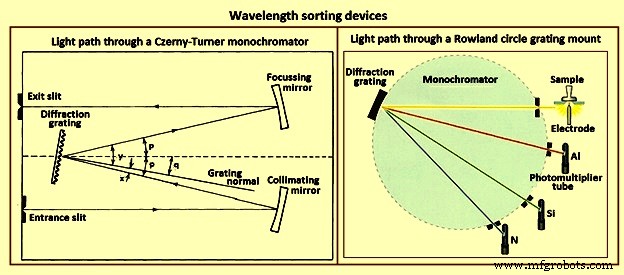
Abb. 4 Wellenlängen-Sortiergerät
Der zweite allgemeine Typ eines Wellenlängensortierers ist der polychromatische. Die meisten Polychromatoren sind Variationen der Rowland-Kreishalterung (Abb. 4). Das Beugungsgitter ist konkav mit einem Krümmungsradius R. Befindet sich ein Eintrittsspalt auf einem Kreis mit Radius R/2, der die Gitterfläche tangiert, werden die Beugungsbilder des Spalts um den Kreis herum fokussiert. Ausgangsschlitze und Photovervielfacherröhren können an Positionen auf der Fokuskurve angeordnet werden, die Wellenlängen von Linien von verschiedenen Elementen entsprechen. Linienintensitäten von 40 bis über 60 Elemente können je nach Geräteleistung gleichzeitig bestimmt werden.
Alternativ kann anstelle der Schlitze und Photomultiplierröhren ein Filmstreifen oder eine Fotoplatte in der Fokuskurve positioniert werden, wodurch der Polychromator in einen Spektrographen umgewandelt wird. Ein ganzes Emissionsspektrum kann in kurzer Zeit auf einer Platte oder einem Stück Film aufgezeichnet werden. Die fotografische Detektion ermöglicht mehr Flexibilität bei der Linienauswahl und liefert mehr Informationen als die Kombination aus festen Schlitzen und Photovervielfacherröhren. Die Zeit, die benötigt wird, um das fotografische Medium zu verarbeiten, die interessierenden Linien zu lokalisieren und ihre Intensität aufzuzeichnen, macht die Verwendung von fotografischen Instrumenten jedoch mühsam. Fortschritte bei den Datenerfassungs- und -verarbeitungsfähigkeiten computergestützter Polychromatoren führen dazu, dass spektrografische Instrumente nicht mehr verwendet werden.
Eine Sammeloptik für ein spektroskopisches Instrument überträgt Strahlungsleistung von der Quelle zum Detektor mit maximaler Effizienz und löst räumliche Heterogenitäten in der Emission von der Quelle auf oder mischt sie in einigen Fällen durcheinander. Die erste Anforderung ist erfüllt, wenn Strahlung von der Quelle den Eintrittsspalt und die Kollimationsoptik des Spektrometers füllt. Eine einfache Linse geeigneter Größe kann verwendet werden, um die Quelle auf dem Eintrittsspalt mit ausreichender Vergrößerung abzubilden, um ihn auszufüllen. Die Größe der Linse wird so gewählt, dass die durch den Schlitz tretende Strahlung gerade die Kollimationsoptik ausfüllt. Der Eintrittsschlitz definiert dann den vom System betrachteten Bereich der Quelle, und jede Quellenungleichförmigkeit innerhalb dieses Bereichs wird auf den Detektor übertragen. Die fotografische Detektion erfordert häufig eine räumliche Gleichmäßigkeit der Spaltbilder. Die gewünschte Gleichmäßigkeit wird erreicht, wenn die Quelle durch eine Linse in der Nähe des Schlitzes auf die Kollimationsoptik abgebildet wird. Andere Linsen werden dann verwendet, um ein Zwischenbild der Quelle bei einer Blende zu erzeugen, um eine räumliche Auflösung bereitzustellen.
Emissionsquellen
Eine Emissionslichtquelle soll die Probe aus einer leicht herzustellenden Form in einen atomaren Dampf zerlegen und dann den Dampf mit ausreichender Effizienz anregen, um ein messbares Emissionssignal von den interessierenden Probenkomponenten zu erzeugen. Jede der vier Arten von Emissionsquellen (Lichtbögen, Hochspannungsfunken, Glimmentladungen und Flammen) hat eine Reihe von physikalischen Eigenschaften mit begleitenden analytischen Vorzügen und Verbindlichkeiten.
Erregungsmechanismen – Die Eigenschaft einer Emissionsquelle, die am engsten mit ihren Anregungseigenschaften verbunden ist, ist die Temperatur. Die Temperatur gibt die Menge an zugänglicher Energie in der Quelle an. Da Energie unterschiedlich auf verschiedene Arten aufgeteilt werden kann, können unterschiedliche Temperaturen diese Aufteilung widerspiegeln. Die kinetische Temperatur des Gases und die Elektronentemperatur geben die kinetische Energie von schweren Teilchen bzw. Elektronen an. Anregungs- und Ionisationstemperaturen spiegeln den elektronischen Energieinhalt von atomaren und molekularen Spezies wider.
Darüber hinaus speichern Moleküle Energie in Rotations- und Vibrationsmodi, die als Vibrations- und Rotationstemperaturen ausgedrückt werden. In vielen Quellumgebungen wird überschüssige Energie in einem Modus schnell zu einem anderen ausgetauscht oder übertragen. In solchen Fällen sind alle oben genannten Temperaturen gleich und die Quelle befindet sich im lokalen thermodynamischen Gleichgewicht (LTE). Wenn LTE existiert, können Anregungsbedingungen ohne Verständnis der mikroskopischen Mechanismen der Energieübertragung beschrieben werden. Die Populationsverteilung unter den möglichen angeregten Zuständen für eine gegebene Spezies ist durch die Boltzmann-Gleichung gegeben.
Wenn LTE nicht existiert, muss eine vollständige Beschreibung der Erregung in solchen Fällen mikroskopische Kollisionsprozesse berücksichtigen, die ein bestimmtes Energieniveau mit einer Effizienz anregen oder abregen können, die sich weit von der unter Verwendung von LTE vorhergesagten unterscheidet. Beispielsweise kann bei Niederdruckentladungen ein kleiner Teil der Elektronenpopulation eine Temperatur haben, die weit über der Gastemperatur in der Entladung liegt. Diese schnellen Elektronen können hoch angeregte Atome oder Ionen in viel größerer Zahl erzeugen, als unter LTE-Bedingungen erzeugt werden. Die Anregungseffizienz in Nicht-LTE-Quellen hängt häufig von engen Übereinstimmungen in der kinetischen oder inneren Energie kollidierender Spezies ab und zeigt daher starke Schwankungen, wenn sich die chemische Zusammensetzung der Anregungsregion ändert.
Ideale Emissionsquelle – Die ideale Emissionsquelle beprobt alle Materialien unabhängig von ihrer Form effizient und liefert Dampf in die Anregungszone mit einer Zusammensetzung, die direkt proportional zur Probenzusammensetzung ist. Die Anregung ist für alle Elemente gleich effizient. Es erzeugt einfache Spektren, wobei die gesamte Anregungsenergie auf wenige angeregte Zustände konzentriert ist. Die Quelle erzeugt kein Hintergrundspektrum. Daher sind die Analyseergebnisse für gleiche Konzentrationen von Elementen in zwei Proben identisch, unabhängig von Unterschieden in der Konzentration anderer Probenbestandteile. Das heißt, Abtasten und Erregen haben keine Matrixabhängigkeit.
Funkenquellen – Der Hochspannungsfunke ist eine intermittierende elektrische Entladung, die durch Betriebsspannungen gekennzeichnet ist, die ausreichen, um einen spontanen Durchbruch einer analytischen Lücke zu verursachen, und hohe Ströme, die aus kapazitiv gespeicherter Energie im Entladungskreis resultieren. Abb. 5 zeigt eine Funkenquelle mit gesteuerter Wellenform, die aus einer Hochspannungs-Ladeschaltung, einem Induktor-Kondensator-Schwingkreis mit einem Hochspannungsschalter und einer Wellenformungsschaltung mit integrierter Analyselücke besteht.
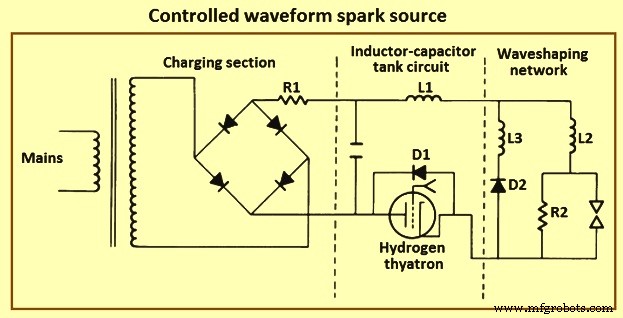
Abb. 5 Funkenquelle mit kontrollierter Wellenform
Die Schaltung erzeugt eine Reihe identischer Funkenentladungen mit präziser Steuerung der Stromstärke und -richtung sowie der Entladungsdauer. In der Praxis ist der Ladeteil der Schaltung einfach ein Hochspannungstransformator und ein Vollwellengleichrichter. Der Funke wird zu Zeiten ausgelöst, die gegenüber dem Nulldurchgang der Ladewellenform des Wechselstroms (AC) verzögert sind, die ausgewählt ist, um zu Beginn jeder Entladung dieselbe Kondensatorspannung zu erzeugen. Der Auslöser ist normalerweise ein Wasserstoff-Thyratron oder ein Hochspannungs-Silizium-gesteuerter Gleichrichter. Für eine gegebene Entladespannung diktieren die relativen Werte der Induktivitäten und der Kapazität die Form und Amplitude der Stromwellenform in den Tank- und Wellenformungsabschnitten der Schaltung. Für den analytischen Betrieb werden die Komponentenwerte normalerweise so ausgewählt, dass sie einen unidirektionalen Entladungsstrom mit Spitzenamplituden von 50 A bis 200 A und Dauern von 50 Mikrosekunden bis 150 Mikrosekunden liefern.
Einschränkungen der Analyse – Die analytische Funkenstrecke besteht typischerweise aus einer Wolframstiftanode und einer Kathode aus dem zu analysierenden Material. Da die Probe eine der Elektroden bildet, ist die funkenemissionsspektroskopische Analyse auf Proben beschränkt, die leitfähig sind oder hergestellt werden können. Die Analyse erfolgt normalerweise in einer inerten Atmosphäre, die in einer geschlossenen Kammer oder als strömender Gasmantel bereitgestellt wird. Wenn sie nicht stabilisiert werden, treffen die einzelnen Funken in einem Zug auf verschiedene Stellen auf der Probenelektrode auf und erzeugen ein mehrere Millimeter breites Brandmuster auf einer ebenen Probe. Mit einem Argonmantel, der von der Anode zur Kathode fließt, schlagen die Funken viel reproduzierbarer und die Brandfläche wird um den Faktor zehn reduziert.
Wenn ein Funke auf die Probenelektrode auftrifft, wird durch schnelle lokale Erwärmung Elektrodenmaterial in die Funkenstrecke geschleudert. Bei einem nicht stabilisierten Funken ist die Flugbahn des ausgestoßenen Materials zufällig. Bei einem stabilisierten Funken breitet sich das Material durch den Spalt als expandierender Zylinder um die Achse zwischen den Elektroden nach oben aus. In jedem Fall wird der Dampf während eines einzigen Funkens verschiedenen Anregungsbedingungen ausgesetzt. Es durchläuft zunächst den energiereichen Kathodenfleck, wo es mehrere Stufen der Ionisation durchlaufen kann. Während er sich weiter nach oben bewegt, bleibt der Dampf in dem stromleitenden Funkenkanal stark erregt, der Dampf, der von der Zwischenelektrodenachse entfernt wird, erfährt viel weniger energetische Bedingungen. Der Strom in der Entladung nimmt zu und ab, wodurch die Anregungsbedingungen deutlich geändert werden, was zeitlich mit der Bewegung des Probendampfs durch den Spalt zusammenfällt.
Die zeitliche und räumliche Inhomogenität des Funkens schließt eine Charakterisierung durch eine Anregungstemperatur aus. Aus mehreren Stufen der Ionisierung des Probenmaterials und des atmosphärischen Gases wird zu unterschiedlichen Zeiten und an unterschiedlichen Orten eine Emission erzeugt. Die dominante Form der Emission ist häufig das erste Ionenspektrum. Linien aus dem einfach geladenen Ion werden traditionell als Funkenlinien bezeichnet.
Änderungen in der Funkenemission treten innerhalb von Mikrosekunden auf. Die Emission einer Funkenkette variiert ebenfalls innerhalb von Minuten. Diese langfristige Änderung der Emissionsintensität, der Sparking-Off-Effekt, ist hauptsächlich eine Widerspiegelung der Änderung in der Probenelektrode, die durch wiederholte Funkenbildung an ihrer Oberfläche verursacht wird. Chemische und physikalische Veränderungen in der Elektrode tragen zum Abfunkeffekt bei. Daher hängt die genaue Art der Zündkurven stark von den experimentellen Bedingungen ab.
Parameter der Funkenquelle (Kapazität, Spannung, Induktivität und Wiederholungsrate), Zusammensetzung der Probe, Phasenstruktur der Probe, Oberflächenbeschaffenheit der Probe, Atmosphäre der Funkenbildung und Brandfläche sind bei der Beschreibung des Funkenbildungsverhaltens zu berücksichtigen. Von besonderer Bedeutung sind die Abhängigkeiten von Probenzusammensetzung und Phasenstruktur, was darauf hindeutet, dass die Emissionsergebnisse für ein Element stark matrixabhängig sind. Dies ist wichtig, wenn der Funke als analytische Emissionsquelle verwendet wird.
Minimierung unerwünschter Eigenschaften – Bei der Funkenanalyse wurden verschiedene Verfahren angewendet, um den Effekt von Quellen-Nicht-Idealitäten zu minimieren. Funkenbildungseffekte werden traditionell behandelt, indem die Emission erst aufgezeichnet wird, nachdem die größten Intensitätsänderungen im Zusammenhang mit einer Verbrennung aufgetreten sind. Das Licht zum Detektor wird während einer Vorbrennperiode blockiert, die typischerweise etwa 1 Minute dauert, während der der Funke die frische Elektrodenoberfläche konditioniert. Wenn der Funke positionsinstabil ist und einen großen Bereich abtastet, bleibt die Emission nach der Vorbrennperiode für die meisten Elemente für die 30 Sekunden, die zum Aufzeichnen eines Emissionsspektrums benötigt werden, ziemlich konstant.
Lagestabile Funken erzeugen im Vergleich zu instabilen Entladungen zeitlich gestauchte Abfunkkurven. Anstatt auf einen stationären Wert anzusteigen, steigt die Emissionsintensität innerhalb weniger Sekunden auf ein Maximum an und fällt dann auf einen relativ niedrigen Wert ab. Die Emission wird mit fortschreitender Verbrennung zunehmend unregelmäßiger. Der Emissionspeak während der ersten 2 Minuten der Verbrennung enthält Informationen über die Konzentration eines Elements in einer Probe und die Art der Matrix, in der das Element gefunden wird.
Eine zusätzliche Kompensation für die Nichtidealität der Funkenquelle ist die Verwendung von Intensitätsverhältnissen statt unmodifizierter Emissionsintensitäten, um die Elementkonzentration anzuzeigen. Die Intensitäten von Linien von Nebenbestandteilen werden mit Intensitäten von einem Hauptbestandteil der Matrix ins Verhältnis gesetzt. Beispielsweise werden in der Stahlanalyse die Linienintensitäten der Legierungselemente mit der Intensität einer Eisenlinie ins Verhältnis gesetzt. Dieses Verfahren gleicht Schwankungen in der Abtast- und Anregungseffizienz von einer Probe zur anderen etwas aus. Es beinhaltet die implizite Annahme, dass Abtastung und Anregung der Referenzkomponente die gleichen Prozesse für die Nebenbestandteile darstellen. Dies kann nicht immer der Fall sein, insbesondere wenn die Probe Einschlüsse enthält, deren Konzentration sich erheblich von der Masse der Probe unterscheidet.
Die obigen Maßnahmen führen zu keinen zufriedenstellenden Analyseergebnissen, es sei denn, das Funkenspektrometer wird unter Verwendung von Standards kalibriert, die der unbekannten Probe in chemischer Zusammensetzung und physikalischer Form genau entsprechen. Laboratorien, die den Funken zur Analyse verwenden, müssen Normensätze für jede Art von zu analysierendem Material haben. Standards für Funkenuntersuchungen sind nicht einfach herzustellen und müssen im Allgemeinen gekauft werden.
Probenahme und Probenvorbereitung
Die Funkenquellenanregung ist die schnellste Methode, um eine Elementaranalyse eines Metalls oder einer Legierung zu erhalten. Proben werden aus flüssigem Stahl, Feinblech, Halbzeugen oder Fertigprodukten entnommen. Geschwindigkeit ist entscheidend in der Stahlindustrie, für die eine Schmelze aus einem Produktionsofen entnommen und analysiert und die Konzentrationen von Legierungselementen innerhalb vorgegebener Bereiche eingestellt werden müssen.
Bei der Probenahme von flüssigem Stahl wird entweder eine Probenahmesonde verwendet oder kleine Mengen flüssigen Stahls, die mit einem Probenahmelöffel gesammelt werden, in eine Gießform gegossen. Die Probe wird gekühlt. Unnötige Teile der Probe werden mit einer Mühle oder einem Cutter entfernt. Wenn keine Spurenelemente untersucht werden, kann eine Mühle oder ein Bandschleifer verwendet werden. Auf der abgekühlten Probe wird eine flache Oberfläche geschliffen oder geschliffen und dann ohne zusätzliche Vorbereitung in eine Funkenquelle gelegt. Um jedoch die Kontamination zu minimieren, muss die Schleifscheibe oder das Schleifband für jede Probe gewechselt werden. Die Analyse wird durchgeführt und die Ergebnisse werden sofort an den Ofen gesendet, wo entsprechende Anpassungen in der Zusammensetzung der Schmelze vorgenommen werden. Die Probenahme und Analyse dauert höchstens ein paar Minuten. Proben, die den Halb- oder Fertigprodukten entnommen werden, müssen einen Durchmesser von mindestens 12 mm haben. Kleine stäbchenförmige Proben können mit speziellen Geräten analysiert werden. Abb. 6 zeigt schematisch die Probenahme, Probenvorbereitung und Analyse durch das optische Emissionsspektroskop.
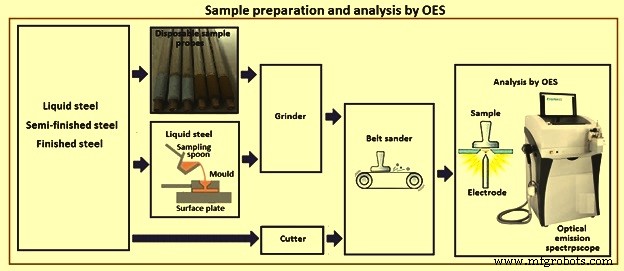
Abb. 6 Probenvorbereitung und Analyse durch OES
Herstellungsprozess
- 16 Schmiedetechniken
- Fotoinduzierte Emissionsanalyse zur Identifizierung von Oberflächenverunreinigungen
- Spektroskopie mit einem optischen Mikroskop
- Metallscheren
- Wirtschaftlichkeit des Blechstanzens
- Was ist Blechstanzen?
- Perforierte Metallmaschine und Anwendungen
- Was ist eine Gießerei?
- Ein Überblick über Dünnblech
- Was ist das Richten von Blech?