


Sensibler nichtenzymatischer elektrochemischer Glukosenachweis basierend auf hohlporösem NiO
Zusammenfassung
Übergangsmetalloxide (TMOs) haben als vielversprechende elektrokatalytische Materialien umfangreiche Forschungsinteressen auf sich gezogen. Trotz geringer Kosten und hoher Stabilität kann die elektrokatalytische Aktivität von TMOs die Anforderungen der Anwendungen noch nicht erfüllen. Inspiriert von der Kinetik gilt das Design einer hohlen porösen Struktur als vielversprechende Strategie, um eine überlegene elektrokatalytische Leistung zu erzielen. In dieser Arbeit wurde eine kubische hohle poröse NiO-Architektur (NiO HPA) durch koordiniertes Ätzen und Präzipitation (CEP)-Prinzip gefolgt von Nachkalzinierung konstruiert. Die NiO-HPA-Elektrode wird zum Nachweis von Glukose eingesetzt und weist eine hervorragende elektrokatalytische Aktivität in Bezug auf eine hohe Empfindlichkeit auf (1323 μA mM −1 cm −2 ) und untere Nachweisgrenze (0,32 μM). Die ausgezeichnete elektrokatalytische Aktivität kann auf eine große spezifische Oberfläche (SSA), geordnete Diffusionskanäle und eine beschleunigte Elektronentransferrate zurückgeführt werden, die von den einzigartigen hohlen porösen Merkmalen herrührt. Die Ergebnisse zeigen, dass der NiO-HPA praktische Anwendungen beim Design von nichtenzymatischen Glukosesensoren haben könnte. Die Konstruktion einer hohlen porösen Architektur bietet eine effektive Nanoengineering-Strategie für Hochleistungselektrokatalysatoren.
Hintergrund
Der Nachweis von Glukose mit einem schnellen, genauen und kostengünstigen Verfahren ist wichtig für die klinische Biochemie, pharmazeutische Analyse, Lebensmittelindustrie und Umweltüberwachung [1,2,3]. Unter den zahlreichen Techniken wurde der elektrochemische Nachweis aufgrund seiner hohen Empfindlichkeit, geringen Kosten und attraktiven unteren Nachweisgrenze als einer der bequemsten Ansätze angesehen [4,5,6]. Die üblichen elektrochemischen Sensoren auf Glucoseoxidase-Basis sind jedoch durch den Nachteil einer unzureichenden Stabilität eingeschränkt, die auf die Natur der Enzyme zurückzuführen ist [7,8,9]. Um diese Probleme anzugehen, wurden aufgrund ihrer geringeren Kosten, physikalisch-chemischen Stabilität und Redox-Elektroaktivität auf TMOs basierte Elektrokatalysatoren empfohlen [10,11,12]. Die gesamte elektrokatalytische Aktivität konventioneller TMOs ist jedoch noch weit von den Anforderungen der Anwendungen entfernt. Es ist immer noch eine Herausforderung, hochaktive TMO-Elektrokatalysatoren für Glucose rational zu entwickeln.
Generell spielt der kinetische Prozess eine entscheidende Rolle für die elektrokatalytische Aktivität etablierter elektrokatalytischer Materialien. Inspiriert von der engen Verbindung zwischen Kinetik und Mikrostruktur kann eine verbesserte elektrokatalytische Aktivität durch das Engineering von Mikrostrukturen, einschließlich Oberfläche, Porenstruktur und Architekturmerkmalen, erreicht werden [13, 14]. Die poröse Struktur bietet eine große spezifische Oberfläche (SSA) und stellt eine Menge aktiver Zentren bereit. Darüber hinaus bietet die poröse Struktur auch genügend Diffusionskanäle für Analyten und Zwischenprodukte, die für den Stofftransportprozess von Vorteil sind [15, 16]. Andererseits können Hohlstrukturen, die funktionale Schalen und innere Hohlräume kombinieren, eine größere Elektrolyt-Elektrode-Kontaktfläche bieten und die Länge sowohl für den Massen- als auch für den Elektronentransport reduzieren [17]. Darüber hinaus verhindern die verfügbaren inneren Hohlräume effektiv die Aggregation elektroaktiver Nanopartikel und nehmen die Volumenänderung und strukturelle Spannung bei wiederholten Messungen auf [18]. Zusammenfassend lässt sich sagen, dass hochaktive TMO-Elektrokatalysatoren durch das Design einer hohlen porösen Architektur erhalten werden können.
Als typisches Übergangsmetalloxid wurde NiO aufgrund des Redoxpaars Ni 3+ . als effizienter Katalysator für die Elektrooxidation von Glucose beschrieben /Ni 2+ im alkalischen Medium, was potenzielle Anwendungen in elektrochemischen Glukosesensoren impliziert. In dieser Arbeit wurde kubischer NiO HPA durch ein Cu2 . konstruiert Koordinierendes Ätzen und Ausfällen (CEP) mit O-Schablone und Nachkalzinierung. Die hohle poröse Struktur bietet große SSA, gut definierte innere Hohlräume, reichlich geordnete Übertragungskanäle und eine hohe Elektronenübertragungseffizienz. Die NiO HPA-Elektrode wird zum Nachweis von Glukose eingesetzt und weist eine höhere Empfindlichkeit und eine niedrigere Nachweisgrenze im Vergleich zu gebrochenem NiO HPA (NiO BHPA) auf, was die Vorteile der hohlen porösen Architektur demonstriert. Diese einfache Strategie zur Konstruktion hohler poröser Architekturen bietet eine gültige Methode zur Entwicklung hocheffizienter Nanomaterialien für elektrochemische Sensoren.
Experimentell
Materialien
CuCl2 ·2H2 O, NiCl2 ·6H2 O, Na2 S2 O3 ·5H2 O, Polyvinylpyrrolidon (PVP, M w =40.000) und NaOH wurden von Chengdu Kelong bezogen. Glucose (Glu.), Lactose (Lact.), Saccharose (Sucr.), Fructose (Fruc.), L-Ascorbinsäure (AA), Harnsäure (UA) und Nafion-Lösung (5 Gew.-% in einer Mischung aus niederen aliphatischen Alkohole und Wasser) wurden ohne weitere Reinigung von Sigma-Aldrich bezogen.
Synthese von Cu2 O-Vorlage
Das kubische Cu2 O-Template wurden gemäß unserer früheren Arbeit [19] synthetisiert. Bei diesem typischen Verfahren wurden 20 ml NaOH (2 M) tropfenweise in 200 ml CuCl2 . gegeben ·2H2 O (10 mM) unter Rühren bei 55 °C. Nach 0,5 h wurden 4 ml AA (0,6 M) tropfenweise in die obige Lösung gegeben. Die Suspension wurde 3 h weiter gealtert und durch Zentrifugation mehrmals mit Wasser gewaschen. Das XRD-Muster sowie die SEM- und TEM-Bilder werden in der zusätzlichen Datei 1 angezeigt:Abbildung S1.
Synthese von NiO HPA
NiO HPA wurde durch ein CEP-Verfahren synthetisiert. Zuerst Cu2 O (10 mg) und NiCl2 ·6H2 O (3 mg) wurden in 10 ml Ethanol-Wasser-Lösungsmittelgemisch (Volumenverhältnis = 1:1) für 7 Minuten durch Ultraschall dispergiert. Dann wurde PVP (0,33 g) unter kräftigem Rühren für 30 Minuten in die Lösung gegeben. Vier Milliliter Na2 S2 O3 (1 M) wurde in das System fallen gelassen; die Reaktion wurde 3 h bei Raumtemperatur fortgesetzt, bis sich die Farbe der Suspension von rot nach hellgrün änderte. Das Ni(OH)2 Vorläufer wurde mehrmals mit warmem Ethanol-Wasser gewaschen und bei Raumtemperatur getrocknet. Schließlich wurde NiO HPA sukzessive unter einer Luftatmosphäre bei 400 °C für 2 h mit einer langsamen Anstiegsrate von 1 °C/min erhalten. NiO BHPA wurde durch starke Ultraschallbehandlung von NiO HPA für 2 Stunden erhalten.
Materialcharakterisierungen
Zusammensetzung und Struktur der Produkte wurden durch Röntgenbeugung (XRD, Rigaku D/Max-2400) charakterisiert. Die Zusammensetzung wurde weiter durch Röntgen-Photoelektronenspektroskopie (XPS, ESCALAB250Xi) mit den C 1s-Peaks bei 284,8 eV als internem Standard analysiert. Die Morphologien und Mikrostrukturen der Produkte wurden mit einem Feldemissions-Rasterelektronenmikroskop (FESEM, FEI Quanta 250, Zeiss Gemini 500) und einem hochauflösenden Transmissionselektronenmikroskop (HRTEM, FEI F20) beobachtet. Brunauer-Emmett-Teller (BET, Belsort-max) wurde verwendet, um die spezifische Oberfläche und Porenstruktur zu analysieren.
Elektrochemische Messungen
Alle elektrochemischen Messungen wurden in 0,1 M NaOH an einer elektrochemischen Workstation von μIII Autolab durchgeführt. Eine Drei-Elektroden-Konfiguration mit NiO HPA (oder NiO BHPA) modifizierter Glaskohlenstoffelektrode (GCE, Ф = 3 mm) als Arbeitselektroden und Ag/AgCl (in gesättigtem KCl) und Platinscheibenelektrode (Ф = 2 mm) als Referenzelektrode bzw. Gegenelektrode. In der Regel wurde GCE mit einer Aluminiumoxid-Aufschlämmung (3, 0,5 und 0,05 μm) poliert. Dann wurde das NiO HPA (10 mg) in einer Mischung aus 0,1 ml Nafion und 0,9 ml destilliertem Wasser gelöst. Schließlich wurden 5 μl der Mischung auf das vorbehandelte GCE getropft (70,77 μg/cm 2 ) und bei Raumtemperatur getrocknet. NiO BHPA-modifiziertes GCE wurde ebenfalls unter den gleichen Bedingungen hergestellt, um die Vorteile von NiO HPA zu verifizieren. Die modifizierten Elektroden wurden durch zyklische Voltammetrie (CV), Chronoamperometrie (CA) und elektrochemische Impedanzspektroskopie (EIS) gemessen, um ihre elektrokatalytische Aktivität zu bewerten. EIS-Messungen wurden über den Frequenzbereich zwischen 0,01 und 100 kHz mit einer Störungsamplitude von 5 mV gegenüber dem offenen Kreispotential durchgeführt.
Ergebnisse und Diskussion
Charakterisierungen
Wie in Abb. 1a gezeigt, entsprechen die Beugungspeaks bei 37,21°, 43,27°, 62,87° und 75,42° (111), (200), (220) und (311) Facetten von kubisch flächenzentriertem NiO ( JCPDS.Nr.47-1049) [20]. Es gibt keine anderen Beugungspeaks, die die Reinheit der Produkte anzeigen. XPS wurde außerdem verwendet, um die Elementzusammensetzung und den Oxidationszustand von NiO HPA zu analysieren. Das Untersuchungsspektrum (Abb. 1b) zeigt O 1s- und Ni 2p-Peaks bei 531,5 bzw. 855,7 eV und enthüllt die Hauptelemente der Produkte. Im Ni 2p-Spektrum (Abb. 1c, siehe Anpassungslinien in Zusatzdatei 1:Tabelle S1), befinden sich zwei Hauptpeaks bei 855,8 eV (Ni 2p3/2 ) und 873,5 eV (Ni 2p1/2 .) ) mit einer Spin-Energie-Trennung von 17,7 eV sind eindeutig untersucht, was das Merkmal der NiO-Phase ist [21]. Die Satellitenpeaks von Ni 2p3/2 und Ni 2p1/2 liegen bei etwa 861,5 bzw. 880,0 eV. Aus Abb. 1d (siehe Anpassungslinien in Zusatzdatei 1:Tabelle S2) ist der passende Peak von O1 bei 529,8 eV die Ni-O-Bindung in Ni-OH-Spezies. Der O2-Peak bei einer Bindungsenergie von 831,3 eV ist normalerweise mit chemisorbiertem Sauerstoff, Hydroxylen und unterkoordiniertem Gittersauerstoff verbunden. Der Peak von O3 bei 532,7 eV ist die Multiplizität von physi- und chemisorbiertem Wasser auf/nahe der Oberfläche [22,23,24]. Die Analyse von XPS und XRD bestätigt die erfolgreiche Herstellung von NiO.

a XRD-Muster von präpariertem NiO-HPA. XPS-Spektren für die Produkte b Umfrage, c Ni 2p und d O 1s
Die Morphologien von Ni(OH)2 Vorläufer (zusätzliche Datei 1:Abbildung S2) und NiO HPA (Abb. 2) wurden durch SEM und TEM deutlich beobachtet. Die SEM-Bilder (Abb. 2a, b) des erhaltenen NiO zeigen ein einheitliches kubisches Merkmal mit einer Kantenlänge von etwa 600 nm. Aus Abb. 2c ist deutlich zu erkennen, dass die raue Hülle von NiO-HPA aus Mengen miteinander verbundener feiner Partikel besteht. Wie in Abb. 2d gezeigt, ist der Rand von NiO-Produkten schwarz und das Innere ist durchscheinend. In Kombination mit den SEM-Beobachtungen in Abb. 2a–c können die kubischen Hohleigenschaften der NiO-Produkte bestätigt werden. Wie in Abb. 2e dargestellt, beträgt die Schalendicke des Würfels etwa 40 nm, was dünner ist als die von Ni(OH)2 . Vorläufer (ca. 60 nm). Die Schrumpfung der Schalendicke wird dem Verlust von H2 . zugeschrieben O im Vorläufer nach der Wärmebehandlung. In Fig. 2f beträgt der Abstand für markierte benachbarte Gitterstreifen etwa 0,21 bzw. 0,24 nm, entsprechend (200)- und (111)-Facetten von NiO. Die Selected Area Electron Diffraktion (SAED)-Ringe können auf (111), (200) und (220) Facetten von NiO innen und außen indiziert werden, was gut mit den XRD-Ergebnissen übereinstimmt [25]. Darüber hinaus zeigen die Elementarabbildungsbilder in Abb. 2g eine oberflächenreiche Verteilung von Ni und O. Wie in Abb. 2h gezeigt, zeigt das Zeilenscan-EDX-Profil die gleichmäßige oberflächennahe Verteilung von O und Ni, was die hohle Architektur erneut bestätigt. NiO-HPA würde genügend aktive Zentren und reichlich Diffusionskanäle bereitstellen, die den Massentransferprozess für Elektrolyt und Glukose begünstigen. Darüber hinaus verkürzt die dünne Hülle von NiO HPA anscheinend die Übertragungsstrecke der Elektronen und beschleunigt die Übertragungsgeschwindigkeit, was NiO HPA eine vielversprechende elektrokatalytische Aktivität verleiht.

a –c SEM und d , e TEM-Bilder von NiO HPA. f Das HRTEM-Bild von NiO HPA. g Die STEM- und EDX-Mapping-Bilder eines NiO-HPA-Würfels. h Die Zeilenscan-EDX-Spektren eines NiO-HPA-Würfels
Um den relevanten Bildungsmechanismus zu verstehen, wurde der nach 0, 10, 20, 30 und 180 min hergestellte Niederschlag gesammelt und durch TEM beobachtet. Wie in Abb. 3a gezeigt, ist das feste kubische Cu2 O-Kristall hat eine Kantenlänge von etwa 600 nm. Mit der Einführung von S2 O3 2− , das koordinierende Ätzen von Cu2 O tritt aufgrund der höheren Diffusionsintensität bevorzugt an der Ecke auf [26]. Wenn die Reaktion fortschreitet, wird das innere Cu2 O-Template schrumpfen signifikant zu einer oktaederähnlichen Struktur, bis sie vollständig entfernt sind. Wie in Abb. 3b zu sehen ist, wird die Farbe des Reaktionssystems allmählich flach und es bilden sich gleichzeitig hellgrüne Niederschläge. In Kombination mit TEM-Ergebnissen wurden die gesamte CEP-Route und der Bildungsmechanismus in Abb. 3c veranschaulicht. Der CEP-Mechanismus kann wie folgt beschrieben werden:(i) Cu + bevorzugt lösliches [Cu2 (S2 O3 2− ) x ] 2 − 2x komplex durch die Kombination mit S2 O3 2− (Reaktion (1)) und gleichzeitig OH − es ist veröffentlicht worden; (ii) Die teilweise Hydrolyse von S2 O3 2− fördert die Versorgung mit OH − (Reaktion (2)). (iii) Die Reaktionen (1) und (2) treiben die Reaktion (3) synchron von links nach rechts voran und erleichtern die Bildung von Ni(OH)2 Schale [27]. In Bezug auf kinetische Faktoren ist die Ätzrate von Cu2 O hängt von der Diffusion von S2 . ab O3 2− vom Äußeren in den Innenraum und die Wachstumsrate von Ni(OH)2 Schale korreliert mit dem Transport von OH − von innen nach außen [28]. Synchrone Steuerung der Ätzrate in Richtung Cu2 O und Ausfällungsrate von Ni(OH)2 Schale führt zur Bildung von wohldefiniertem hohlem Ni(OH)2 Vorläufer. NiO HPA wird schließlich durch die Nachkalzinierung von Ni(OH)2 . gewonnen Vorläufer.
$$ {\mathrm{Cu}}_2\mathrm{O}+x{\mathrm{S}}_2{\mathrm{O}}_3^{2-}+{\mathrm{H}}_2\mathrm{ O}\to {\left[{\textrm{Cu}}_2{\left({\textrm{S}}_2{\textrm{O}}_3\right)}_x\right]}^{2-2x }+2{\mathrm{O}\mathrm{H}}^{-} $$ (1) $$ {\mathrm{S}}_2{\mathrm{O}}_3^{2-}+{\ mathrm{H}}_2\mathrm{O}\rightleftharpoons {\mathrm{H}\mathrm{S}}_2{\mathrm{O}}_3^{2-}+{\mathrm{O}\mathrm{H }}^{-} $$ (2) $$ {\textrm{Ni}}^{+}+2{\textrm{OH}}^{-}\to \textrm{Ni}{\left(\textrm {OH}\right)}_2 $$ (3)
a TEM-Bilder der Produkte, die bei verschiedenen Reaktionszeiten überwacht wurden. b Optische Aufnahmen der Suspension bei unterschiedlicher Reaktionszeit nach Zugabe von Ätzmittel. c Schematische Darstellung des vorgeschlagenen Wachstumsmechanismus von NiO HPA
Die Oberfläche und Porosität von NiO HPA und NiO BHPA (Zusatzdatei 1:Abbildung S3) wurden ebenfalls durch die BET-Methode charakterisiert. NiO HPA besitzt eine SSA von 27,08 m 2 /g und ein Porenvolumen von 0,087 cm 3 /g (Abb. 4a), was viel größer ist als bei den berichteten NiO-Materialien [29]. In Bezug auf die Porengrößenverteilung weist NiO HPA hauptsächlich eine konzentrierte Verteilung bei etwa 7 nm auf, die mit den geordneten Kanälen zwischen NiO-Nanopartikeln zusammenhängt. Die große SSA und die geordneten Kanäle können die Absorption des Analyten und den Massentransportprozess effektiv verbessern, was zu einer erhöhten elektrokatalytischen Aktivität führt. Die SSA und das Porenvolumen der gebrochenen Probe betragen 5,24 m 2 /g und 0,078 cm 3 /g (Abb. 4b), die viel kleiner ist als die von NiO HPA. Dies ist auf den Zusammenbruch der ursprünglichen Hohlstruktur nach der Ultraschallbehandlung zurückzuführen. Bemerkenswert ist, dass für NiO BHPA keine konzentrierte Porenverteilung beobachtet wird (Einschub von Fig. 4b), was auf eine vollständige Zerstörung der geordneten Diffusionskanäle hinweist. Die Abnahme von SSA und die Zerstörung geordneter Diffusionskanäle sind für die Kinetik ungünstig, was zu einer schlechten elektrokatalytischen Aktivität führen kann. Dementsprechend besitzt NiO HPA im Vergleich zu zerbrochenen Proben vorteilhafte Mikrostrukturen für die Elektrokatalyse.

N2 Adsorptions-Desorptions-Isothermen von a NiO HPA und b NiO-BHPA. Einschub von a und b sind die entsprechenden Porengrößenverteilungen
Elektrochemische Leistung
Abbildung 5a zeigt die CVs von NiO-HPA- und NiO-BHPA-Elektroden mit und ohne 1 mM Glukose. Ein Paar gut definierter Peaks bei 0,48 und 0,38 V wird in Kurve III deutlich untersucht, die sich auf Ni 2+ . beziehen /Ni 3+ Redox-Paar. Der Redox-Spitzenstrom von Kurve III ist offensichtlich höher als der von Kurve I. Dies hängt mit dem Zusammenbruch der Hohlarchitektur und der Abnahme von SSA zusammen. Bei Zugabe von Glucose werden Stromreaktionen an beiden Elektroden deutlich beobachtet (Kurve II und IV). Die NiO-HPA-Elektrode zeigt eine höhere Stromantwort als die NiO-BHPA-Elektrode. Darüber hinaus ist das Einsetzpotential für die Elektrooxidation von Glucose an der NiO-HPA-Elektrode (0,43 V) niedriger als das der NiO-BHPA-Elektrode (0,46 V), was eine höhere elektrokatalytische Aktivität zeigt. Die hohe elektrokatalytische Aktivität wird großen Mengen an aktiven Zentren, einer geordneten Porenstruktur und einer hohen Elektronenübertragungsrate zugeschrieben, die durch die hohle poröse Struktur bereitgestellt wird. Die Elektrooxidation von Glucose auf der NiO-HPA-Elektrode wird durch Ni 2+ . angetrieben /Ni 3+ Redoxpaar im alkalischen Medium nach folgenden Reaktionen [30, 31]:
$$ \mathrm{NiO}\to {\mathrm{Ni}}^{2+}+{\mathrm{O}}^{2-} $$ (4) $$ {\mathrm{Ni}}^{ 2+}+{\mathrm{OH}}^{-}\to {\mathrm{Ni}}^{3+}+{e}^{-} $$ (5) $$ {\mathrm{Ni} }^{3+}+\mathrm{Glukose}\to {\mathrm{Ni}}^{2+}+\mathrm{gloconic}\ \mathrm{Säure} $$ (6)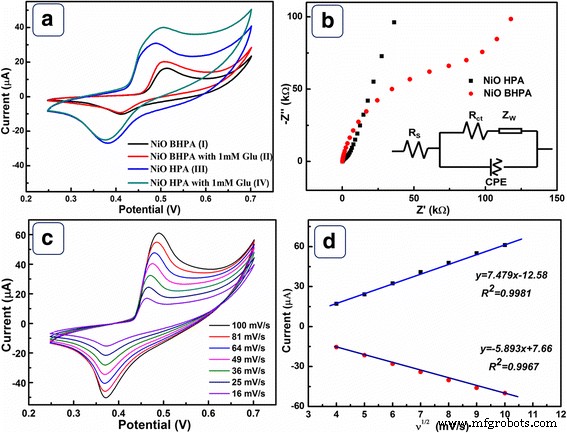
a CVs der NiO BHPA (I, II) und NiO HPA (III, IV) Elektrode mit (II, IV) und ohne (I, III) der Anwesenheit von 1 mM Glukose in 0,1 M NaOH bei einer Scanrate von 50 mV/s. b Nyquist-Diagramme EIS und Ersatzschaltbild von NiO HPA und NiO BHPA in 0,1 M NaOH-Lösung. c CVs der NiO-HPA-Elektrode bei verschiedenen Scanraten in 0,1 M NaOH mit 1 mM Glukose und d die Beziehung zwischen Spitzenstrom und Quadratwurzel der Abtastraten
Wie oben gezeigt, OH − spielt eine wichtige Rolle bei der elektrokatalytischen Reaktion. Offensichtlich beschleunigt alkalisches Medium das Redox von Ni 2+ /Ni 3+ im Vergleich zum neutralen Medium (zusätzliche Datei 1:Abbildung S4), was zu einer höheren elektrokatalytischen Aktivität führt.
Nyquist-Plots von NiO HPA- und NiO BHPA-Elektroden wurden in Abb. 5b dargestellt. Jeder Plot ist im Hochfrequenzbereich durch einen Halbkreis und im Niederfrequenzbereich durch eine Gerade gekennzeichnet. Im Allgemeinen repräsentiert der Achsenabschnitt auf der reellen Achse den Lösungswiderstand (R s ), die sich aus Eigenwiderstand, Ionenwiderstand und Kontaktwiderstand zusammensetzt. Der Halbkreisdurchmesser in Bezug auf den Elektronenübergangswiderstand wird durch R . dargestellt ct . Wie in Zusatzdatei 1:Tabelle S3 gezeigt, weist die NiO-HPA-Elektrode ein kleineres R . auf s und R ct als NiO-BHPA. Die Tatsachen können der vorteilhaften Elektronentransferkinetik zugeschrieben werden, die sich aus dem hohlen Merkmal ergibt. Die Steigung der Impedanzkurve im Niederfrequenzbereich entspricht der Warburg-Impedanz (Z w ), der den Diffusionswiderstand darstellt [32]. Es ist klar, dass NiO HPA die Diffusionskinetik begünstigt; jedoch behindert das NiO BHPA die Diffusion des Elektrolyten. Dies ist auf die Zerstörung der geordneten Diffusionskanäle nach Ultraschall zurückzuführen. Auf der Grundlage der obigen EIS-Diskussionen ist die NiO HPA-Elektrode im Vergleich zur zerbrochenen Probe sowohl für die Elektronen- als auch für die Massentransferkinetik vorteilhafter, was die Vorteile von NiO HPA als Elektrokatalysator für Glukose impliziert.
Die Kinetik der NiO-HPA-Elektrode wurde aus den CVs mit unterschiedlichen Scanraten in 1 mM Glucoselösung bestimmt (Abb. 5c). Wie in Abb. 5d dargestellt, sind die anodischen und kathodischen Spitzenströme proportional zur Quadratwurzel der Abtastraten, was einen typischen diffusionsgesteuerten dynamischen Prozess demonstriert. Darüber hinaus wird keine signifikante positive/negative Verschiebung für den anodischen/kathodischen Peak beobachtet, was auf eine ungehinderte Diffusionskinetik hinweist, die von der hohlen porösen Struktur stammt.
Die Selektivität, Reproduzierbarkeit und Stabilität der NiO-HPA-Elektrode
Um ein optimiertes Arbeitspotenzial zu erhalten, wurden die aktuelle Reaktion von Glukose und die Interferenz von AA bei verschiedenen Potenzialen berücksichtigt und die Daten in Abb. 6a dargestellt. Aus den statistischen Daten in Abb. 6b wurde 0,6 V aufgrund der Tatsache ausgewählt, dass die NiO-HPA-Elektrode bei 0,6 V eine maximale Stromantwort auf Glukose und eine minimale Störung auf AA zeigt Glukosekonzentration bei 0,6 V. Bemerkenswerte Stromreaktionen werden für die beiden Elektroden deutlich beobachtet, und die Stromreaktionen nehmen mit steigender Glukosekonzentration zu. Abbildung 6d zeigt die Beziehung zwischen Ansprechströmen und Glukosekonzentration für NiO-HPA- und NiO-BHPA-Elektroden. Die NiO-HPA-Elektrode bietet einen linearen Bereich von 0,32 bis 1100 μM mit einer Empfindlichkeit von 1323 μA mM −1 cm −2 , die höher ist als die der NiO-BHPA-Elektrode (753 μA mM −1 cm −2 ). Darüber hinaus ist die Nachweisgrenze (LOD) der NiO-HPA-Elektrode (0,32 μM) viel niedriger als die der NiO-BHPA (14,2 μM). Um die Vorteile von NiO HPA zu zeigen, wurde die Leistung der NiO HPA-Elektrode mit anderen berichteten NiO-basierten Glucose-Detektionselektroden in Tabelle 1 verglichen. Es wurde festgestellt, dass die NiO-HPA-Elektrode eine zufriedenstellende elektrokatalytische Aktivität gegenüber Glucose in Bezug auf hohe Empfindlichkeit und niedrige LOD . aufweist , was auf mögliche Anwendungen in elektrochemischen Glukosesensoren hinweist. Dies wird im Wesentlichen den zahlreichen aktiven Zentren, der schnelleren Massentransportkinetik und der beschleunigten Elektronentransferkinetik zugeschrieben, die von der hochporösen Hohlarchitektur abgeleitet werden.

a Amperometrische Reaktion der NiO-HPA-Elektrode bei verschiedenen Potentialen unter Zugabe von 0,1 mM Glukose und 0,01 mM AA. b Der Reaktionsstrom von Glukose und AA bei unterschiedlichen Potentialen. c CA der NiO HPA- und NiO BHPA-Elektrode bei 0,6 V mit sukzessiver Zugabe von Glucose. d Die Beziehung zwischen Reaktionsstrom und der Glukosekonzentration
Die Selektivität ist ein wichtiger Indikator zur Beurteilung der Leistung von Glukosesensoren. Einige leicht oxidierbare Verbindungen, wie Lact., Sucr., Fruc., UA und AA, existierten normalerweise zusammen mit Glucose im menschlichen Blut. Bemerkenswerterweise beträgt der physiologische Spiegel dieser störenden Spezies etwa ein Zehntel der Glukosekonzentration [33]. Daher wurde die Selektivität der NiO-HPA-Elektrode bewertet, indem während der amperometrischen Messung 0,01 mM oberhalb der störenden Spezies in Richtung 0,1 mM Glukose eingeführt wurden. Wie in Abb. 7a gezeigt, werden für Lact., Sucr., Fruc. und UA keine schwerwiegenden Interferenzen beobachtet. Die Hauptstörspezies AA zeigt nur 8,7% Störstrom gegenüber Glucose. Darüber hinaus behält die zweite Zugabe von 50 μM Glukose noch etwa (89 ± 0,2) % ihrer ursprünglichen Reaktion bei, was auf eine hervorragende Anti-Interferenz-Leistung hinweist. Die herausragende Selektivität könnte dem elektrostatischen Abstoßungseffekt zwischen der NiO-HPA-Elektrode und störenden Spezies zugeschrieben werden. Die NiO-HPA-Elektrode wäre in 0,1 M NaOH negativ geladen, da der pH-Wert des Elektrolyten über dem isoelektrischen Punkt von NiO liegt [34]. Darüber hinaus verliert die Hauptinterferenzspezies (AA) in alkalischer Lösung leicht Protonen und besitzt eine negativ geladene Hülle [35]. Die elektrostatische Abstoßung zwischen der Hülle der Störsubstanz und der NiO-HPA-Elektrode führt zu einer verbesserten Selektivität. Die Stabilität der NiO-HPA-Elektrode wurde durch Messung ihrer aktuellen Reaktion auf 0,1 mM Glukose über 30 Tage geschätzt. In Abb. 7b behält die aktuelle Reaktion nach 30 Tagen noch 83,13 % ihrer ursprünglichen Reaktion bei, was eine ausgezeichnete Langzeitstabilität der NiO-HPA-Elektrode bei Raumtemperatur zeigt. Die aktuelle Reaktion der NiO-HPA-Elektrode auf 0,1 mM Glukose ist über eine Betriebszeit von 2000 s stabil, mit einem Verlust von 9,82 % ihrer ursprünglichen Reaktion. Die fünf unabhängig voneinander hergestellten NiO-HPA-Elektroden zeigen eine akzeptable RSD von 3,12 % für Stromreaktionen gegenüber 0,1 mM Glukose bei 0,6 V. Darüber hinaus wurden Stromreaktionen für dieselbe NiO-HPA-Elektrode gegenüber 0,1 mM Glukose zehnmal gemessen und die Stromreaktionen zeigen a RSD von 2,36 %, was eine bemerkenswerte Reproduzierbarkeit zeigt. Die NiO-HPA-Elektrode zeichnet sich durch hohe Empfindlichkeit, ausgezeichnete Stabilität und bemerkenswerte Reproduzierbarkeit aus, was sie für praktische Anwendungen attraktiv macht.

a Die Stromantwort der NiO-HPA-Elektrode auf die sequentielle Zugabe von 50 μM Glukose und 5 μM störender Spezies bei einem angelegten Potenzial von 0,6 V. Einschub sind die statistischen Daten des Interferenzstroms. b Langzeitstabilität der NiO-HPA-Elektrode für 0,1 mM Glukose. Eingesetzt ist die Stabilität der NiO-HPA-Elektrode mit der Laufzeit. c Aktuelle Reaktionen von fünf NiO-HPA-Elektroden auf 0,1 mM Glukose. d Zehn Messungen einer NiO-HPA-Elektrode gegen 0,1 mM Glukose
Nachweis von Glukose in Humanserum
Darüber hinaus wurde eine NiO-HPA-Elektrode verwendet, um den Glukosespiegel im menschlichen Blut zu bestimmen, und die Ergebnisse wurden mit einem medizinischen Gerät verglichen (Tabelle 2). Die Serumproben wurden von einem örtlichen Krankenhaus bereitgestellt und vor den Messungen mit alkalischen Elektrolyten verdünnt [36, 37]. Der bei 0,6 V gemessene Reaktionsstrom wurde aufgezeichnet, um die entsprechende Glukosekonzentration gemäß der Arbeitsgleichung zu berechnen. Die NiO-HPA-Elektrode zeigt eine RSD von 2,85% für den Nachweis von Glukose. Darüber hinaus weist die NiO-HPA-Elektrode eine akkreditierte Wiederfindung zwischen 92 und 102 % auf, was eine ausgezeichnete Praktikabilität bei der Bestimmung von Glukose in Humanserum beweist.
Schlussfolgerungen
Zusammenfassend haben wir mit einer CEP-Methode erfolgreich einen NiO-HPA-Elektrokatalysator für Glucose hergestellt. Der NiO-HPA bietet große SSA, geordnete Porenstruktur und kurze elektronische Übertragungswege, die für die elektrokatalytische Kinetik von Vorteil sind. Als nicht-enzymatische Glukose-Detektionselektrode weist NiO HPA eine höhere Empfindlichkeit von 1323 μA mM –1 . auf cm −2 und eine niedrigere LOD von 0,32 μM im Vergleich zu NiO-BHPA. Im Hinblick auf die Selektivität werden weniger als 8,7 % Interferenz für die üblichen störenden Spezies untersucht. Gleichzeitig behält die NiO-HPA-Elektrode nach 30 Tagen 89,02 % ihrer ursprünglichen Reaktion bei. Darüber hinaus wurde der entwickelte NiO-HPA erfolgreich zum Nachweis von Glukose in Humanserum eingesetzt. NiO HPA bietet im Vergleich zu medizinischen Geräten eine akkreditierte Stabilität und Praktikabilität. Das Design einer hohlen porösen Architektur ebnet einen hocheffizienten Weg, um kostengünstige und leistungsstarke Elektrokatalysatoren für Glukose zu erhalten.
Nanomaterialien
- 8051 Mikrocontroller-basierter Ultraschall-Objekterkennungsschaltkreis
- Demonstration eines flexiblen Graphen-basierten Biosensors für den empfindlichen und schnellen Nachweis von Eierstockkrebszellen
- Ein hochempfindlicher elektrochemischer DNA-Biosensor aus Acryl-Gold-Nanokomposit zur Bestimmung des Geschlechts von Arowana-Fischen
- Mesoporöse Nickeloxid (NiO)-Nanoblätter für die hochempfindliche Glukosemessung
- Hochempfindlicher chemischer Ethanolsensor basierend auf neuartigem Ag-dotiertem mesoporösem α-Fe2O3, hergestellt durch modifiziertes Sol-Gel-Verfahren
- Schichtpassiviertes poröses Silizium aus Graphen mit wenigen Schichten für eine hervorragende elektrochemische Doppelschicht-Superkondensatorelektrode
- Hochselektiver und empfindlicher Nachweis von Hg2+ basierend auf Förster-Resonanzenergietransfer zwischen CdSe-Quantenpunkten und g-C3N4-Nanoblättern
- Rationales Design einer hohlporigen Ni(OH)2-Architektur für einen hochempfindlichen enzymfreien Glukosesensor
- Ein neuartiger magnetoelastischer Nanobiosensor für den hochempfindlichen Nachweis von Atrazin
- Plasmonischer ELISA zum sensiblen Nachweis von Krankheitsbiomarkern mit einem Smartphone-basierten Lesegerät