


Analyse durch Röntgenfluoreszenzspektrometrie
Analyse durch Röntgenfluoreszenzspektrometrie
Röntgenfluoreszenz (XRF) ist eine emissionsspektroskopische Technik, die auf dem Gebiet der Elementidentifikation und -bestimmung breite Anwendung gefunden hat. Die Technik hängt von der Emission charakteristischer Röntgenstrahlung ab, normalerweise im Energiebereich von 1 keV bis 60 keV, nach Anregung von atomaren Elektronenenergieniveaus durch eine externe Energiequelle, wie beispielsweise einen Elektronenstrahl, einen Strahl geladener Teilchen oder ein x - Strahl. In den meisten Probenmatrizes kann die Röntgenspektrometrie Elemente in Konzentrationen von weniger als 1 Mikrogramm/g Probe (1 ppm) nachweisen. In einer Dünnfilmprobe kann es Gesamtmengen von wenigen Zehntel Mikrogramm nachweisen. Anfänglich fand die Röntgenspektrometrie breite Akzeptanz in Anwendungen im Zusammenhang mit metallurgischen und geochemischen Analysen. In jüngerer Zeit hat sich die Röntgenspektrometrie als wertvoll bei der Analyse von Umweltproben, bei der Bestimmung von Schwefel und Verschleißelementen in Erdölprodukten, bei Anwendungen mit forensischen Proben und bei Messungen von elektronischen und computerbezogenen Materialien erwiesen.
Röntgenfluoreszenz (XRF)-Spektrometrie ist ein vielseitiges Werkzeug für viele analytische Probleme. Haupt-, Neben- und Spurenelemente können in verschiedenen Arten von Proben wie Metallen, Legierungen, Gläsern, Zementen, Mineralien, Gesteinen, Erzen, Polymeren sowie Umwelt- und biologischen Materialien qualitativ und quantitativ bestimmt werden. Elemente von Natrium (Na) bis Uran (U) werden routinemäßig mit einem energiedispersiven Röntgenfluoreszenz-Spektrometer (EDXRF) bestimmt, während die Anwendung eines wellenlängendispersiven Röntgenfluoreszenz-Spektrometers (WDRFA) eine effiziente Bestimmung von Low-Z-Elementen bis hinunter zu ermöglicht sogar Beryllium (Be). Obwohl die Proben ohne Behandlung analysiert werden können, können bei entsprechender Probenvorbereitung qualitativ hochwertige Ergebnisse sichergestellt werden. Dies kann vom einfachen Reinigen und Polieren der Probe (Metalle, Legierungen), Pulverisieren und Pelletieren mit oder ohne Bindemittel (Keramik, Mineralien, Erze, Böden usw.) etc.) zum Aufschluss mit Säuren (Metalle, Legierungen). Auf diese Weise können Fehler, die durch Oberflächenrauheit, Partikelgrößeneffekt oder Inhomogenität des Materials entstehen, eliminiert oder minimiert werden.
Röntgen entdeckte 1895 die Röntgenstrahlen. H.G.J. Moseley entwickelte die Zusammenhänge zwischen Atomstruktur und Röntgenemission und veröffentlichte 1913 die ersten Röntgenspektren, die die Grundlage der modernen Röntgenspektrometrie bilden. Moseley erkannte das Potenzial für quantitative Elementbestimmungen unter Verwendung von Röntgentechniken. In den folgenden Jahrzehnten erfolgte die Entwicklung routinemäßiger Röntgengeräte, die zum heute bekannten Röntgenspektrometer führten. Coolidge entwarf 1913 eine Röntgenröhre, die den derzeit verwendeten ähnlich ist. Soller erreichte 1924 die Kollimation von Röntgenstrahlen. Verbesserungen des Gasröntgendetektors von Geiger und Mueller im Jahr 1928 führten schließlich 1948 zum Design des ersten kommerziellen WDXRF von Friedman und Birks. In jüngerer Zeit wurden andere Detektoren, wie z Germanium und die mit Lithium dotierten Silizium-Halbleiterdetektoren haben zu modifizierten Konstruktionen von Röntgenspektrometern geführt. Moderne energiedispersive Instrumente erleichtern die qualitative Identifizierung von Elementen in verschiedenen Proben. Der Informationsgehalt eines energiedispersiven Röntgenspektrums gehört zu den höchsten, die von anorganischen Materialien in einer einzigen Messung erhalten werden können. Die Position und Intensität der spektralen Peaks liefern qualitative und quantitative Informationen, und die Intensität des Hintergrunds liefert Informationen über die Massenzusammensetzung der Probenmatrix.
Die Röntgenspektrometrie ist eine der wenigen Techniken, die auf feste Proben unterschiedlicher Form angewendet werden kann. Obwohl die meisten XRF-Spektrometer in Labors verwendet werden, finden viele Anwendung in Routineanalysen für die Produktions- und Qualitätskontrolle und in speziellen Aufgaben. Ein Strukturdiagramm des EDXRF-Spektrometers ist in Abb. 1 dargestellt.
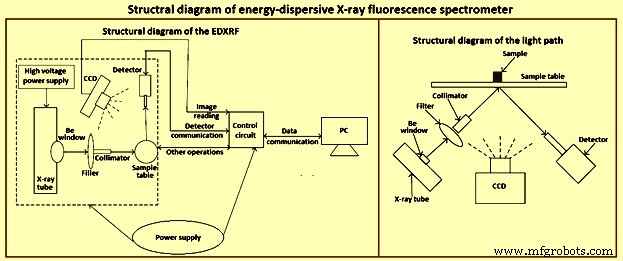
Abb. 1 Strukturdiagramm des EDXRF-Spektrometers
Elektromagnetische Strahlung
Elektromagnetische Strahlung ist eine Energieform, die sich durch den Weltraum ausbreiten und mit Atomen und Molekülen interagieren kann, um deren Energiezustand zu ändern. Beide Eigenschaften sind für die Spektroskopie wichtig. Elektromagnetische Strahlung zeigt ein Verhalten, das zwei Theorien zur Erklärung benötigt. Die Wellentheorie beschreibt das Verhalten elektromagnetischer Strahlung, wie Brechung, Reflexion, Beugung und Streuung. Strahlung ist definiert als eine Energieform, die aus zwei orthogonalen Wellen besteht, die jeweils die gleiche Frequenz und Wellenlänge haben. Das eine ist ein oszillierendes elektrisches Feld und das andere ein oszillierendes magnetisches Feld, wodurch der Begriff elektromagnetische Strahlung entsteht.
Im Vakuum ist die Ausbreitungsgeschwindigkeit der Welle durch den Raum die Lichtgeschwindigkeit (c =3 × 10 hoch 10 cm/s). Dies führt zu einer wichtigen grundlegenden Beziehung, die durch die Gleichung w.v =c dargestellt wird. Dieser Ausdruck besagt, dass das Produkt aus der Wellenlänge (w) der elektromagnetischen Strahlung und ihrer Frequenz (v) gleich ihrer Geschwindigkeit ist. Die Wellenlänge elektromagnetischer Strahlung variiert über viele Größenordnungen. Zum Beispiel haben Funkwellen im normalen AM-Rundfunkband Wellenlängen von mehreren hundert Metern und ultraviolette Wellenlängen liegen im Bereich von 10 nm bis 100 nm (Nanometer). Im Gegensatz dazu reichen Röntgenstrahlen, die in der Spektroskopie nützlich sind, von 0,01 nm bis 10 nm (Abb. 2).

Abb. 2 Röntgenstrahlen und andere elektromagnetische Strahlungen
Für die wellenlängendispersive Spektrometrie ist es oft bequemer, Wellenlängeneinheiten zu verwenden, aber für die energiedispersive Röntgenspektrometrie (EDS) ist die Energiebeschreibung bequemer. Die Interkonvertierung ist jedoch einfach.
Mehrere normalerweise verwendete Beschreibungen der Eigenschaften von Röntgenstrahlen sind bedeutsam. Die eigentliche Bedeutung der Intensität elektromagnetischer Strahlung ist die Energie pro Flächeneinheit pro Zeiteinheit; jedoch wird häufig die Anzahl der Zählungen pro Zeiteinheit vom Detektor als Intensität verwendet. Da die Fläche die aktive Fläche des verwendeten Detektors ist und die Zeit ein einstellbarer Parameter ist, ist die Verwendung von Zählwerten eine praktische Beschreibung der Röntgenstrahlintensität. Die Begriffe harte oder weiche Röntgenstrahlen werden häufig verwendet, um Röntgenstrahlen mit kurzen (0,01 nm bis 0,1 nm) bzw. langen (0,1 nm bis 1 nm) Wellenlängen zu unterscheiden. Röntgenstrahlung fällt in den hochenergetischen Bereich des elektromagnetischen Spektrums.
Röntgenemission
Röntgenstrahlen werden durch die Störung der Elektronenorbitale von Atomen erzeugt. Dies kann auf verschiedene Arten erfolgen, wobei die häufigste die Beschießung eines Zielelements mit hochenergetischen Elektronen, Röntgenstrahlen oder beschleunigten geladenen Teilchen ist. Die ersten beiden werden häufig direkt oder indirekt in der Röntgenspektrometrie verwendet. Der Elektronenbeschuss führt zu einem Kontinuum von Röntgenenergien sowie einer für das Zielelement charakteristischen Strahlung. Beide Strahlungsarten werden in der Röntgenspektrometrie angetroffen.
Kontinuum – Die Emission von Röntgenstrahlen mit einer glatten, kontinuierlichen Funktion der Intensität relativ zur Energie wird als Kontinuums- oder Bremsstrahlung bezeichnet. Ein Röntgenkontinuum kann auf verschiedene Weise erzeugt werden. Am nützlichsten ist jedoch der Elektronenstrahl, der verwendet wird, um ein Ziel in einer Röntgenröhre zu bombardieren. Das Kontinuum wird als Ergebnis der fortschreitenden Verzögerung hochenergetischer Elektronen erzeugt, die auf ein Ziel auftreffen, das eine Verteilung von Orbitalelektronen verschiedener Energien ist. Da die auftreffenden Elektronen mit den gebundenen Orbitalelektronen interagieren, wird ein Teil ihrer kinetischen Energie in Strahlung umgewandelt. Die umgesetzte Menge hängt von der Bindungsenergie des beteiligten Elektrons ab. Daher besteht eine einigermaßen statistische Wahrscheinlichkeit dafür, wie viel Energie bei jeder Wechselwirkung umgewandelt wird.
Die Wahrscheinlichkeit, dass ein auftreffendes Elektron mit einem Orbitalelektron des Zielelements wechselwirkt, steigt mit der Ordnungszahl des Elements, somit steigt die Intensität der Kontinuumemission mit der Ordnungszahl des Zielelements. Außerdem steigt die Wahrscheinlichkeit einer Wechselwirkung mit der Anzahl der Elektronen pro Zeiteinheit im Strahl oder Fluss. Daher steigt die Intensität des Kontinuums mit dem Elektronenstrahlstrom, ausgedrückt in Milliampere. Darüber hinaus steigt die Fähigkeit der auftreffenden Elektronen, mit fest gebundenen Elektronen des Zielelements in Wechselwirkung zu treten, mit der kinetischen Energie der auftreffenden Elektronen. Da die kinetische Energie der Elektronen im Strahl mit dem Beschleunigungspotential zunimmt, steigt die integrierte Intensität des Kontinuums mit dem Elektronenbeschleunigungspotential, ausgedrückt in Kilovolt. Schließlich entspricht die maximale Energie, die sich als Röntgenphotonen manifestiert, der kinetischen Energie des auftreffenden Elektrons, die wiederum mit dem Beschleunigungspotential zusammenhängt. Die Energie der maximalen Intensität im Kontinuum liegt bei etwa zwei Dritteln der maximal emittierten Energie. Ferner gibt es die Absorption von Röntgenstrahlen innerhalb des Zielmaterials oder Absorption durch Materialien, die für Fenster in der Röntgenröhre und den Detektoren verwendet werden. Daher kann eine gewisse Modifikation der Intensitätsverteilung auftreten, insbesondere bei niedrigen Röntgenenergien.
Charakteristische Emission – Die meisten Elektronen, die auf ein Ziel auftreffen, interagieren mit den Orbitalelektronen des Zielelements in unspezifischen Wechselwirkungen und führen zu einer geringen oder keiner Störung der inneren Orbitalelektronen. Einige Wechselwirkungen führen jedoch zum Ausstoß von Elektronen aus diesen Orbitalen. Die resultierenden Leerstellen oder Löcher repräsentieren hochenergetische instabile Zustände. Wenn sich die Orbitallücken in den innersten Schalen befinden, kaskadieren Elektronen von den äußeren Schalen, um sie zu füllen, und dies führt zu einer niedrigeren Energie und einem stabileren Zustand. Die durch den Prozess freigesetzte Energie kann als Röntgenstrahlen manifestiert werden. Jeder der auftretenden Übergänge führt zur Emission scharfer Röntgenlinien, die für das Zielelement und den beteiligten Übergang charakteristisch sind. Diese charakteristischen Strahlungslinien werden mit dem Kontinuum emittiert.
Röntgenabsorption
Auf eine Probe auftreffende Röntgenstrahlen erfahren zwei wichtige Wechselwirkungen mit den Elementen der Probe:Absorption und Streuung. Die Absorption der Strahlung kann durch spezifische Wechselwirkungen erfolgen, die bei der Probenanregung in der Röntgenspektrometrie erheblich sind, oder durch allgemeinere Wechselwirkungen, die die von der Probe emittierte Röntgenintensität beeinflussen. Streuung von Röntgenstrahlen führt zu Hintergrundintensität in den beobachteten Spektren.
Massenaufnahme – Wenn ein Röntgenstrahl ein Material durchdringt, können die Photonen (elektromagnetische Felder) auf unspezifische Weise mit Elektronen in den Orbitalen der Zielelemente wechselwirken, wodurch die Intensität des Röntgenstrahls verringert wird. Die Wechselwirkungen können zu einem photoelektrischen Ausstoß von Elektronen oder einer Streuung des Röntgenstrahls führen. In beiden Fällen wird das Gesamtergebnis häufig als exponentielle Intensitätsabnahme mit der Weglänge des absorbierenden Materials beschrieben. Der Massenabsorptionskoeffizient ist charakteristisch für ein bestimmtes Element bei bestimmten Energien der Röntgenstrahlung. Sein Wert variiert mit der Wellenlänge der Röntgenstrahlung und der Ordnungszahl des Zielelements.
Der photoelektrische Effekt ist der wichtigste Prozess, der zur Absorption von Röntgenstrahlen führt, wenn sie Materie durchdringen. Der photoelektrische Effekt ist der Ausstoß von Elektronen aus den Orbitalen von Elementen im Röntgentarget. Dieser Prozess trägt häufig hauptsächlich zur Absorption von Röntgenstrahlen bei und ist die Art der Anregung der Röntgenstrahlenspektren, die von Elementen in Proben emittiert werden. Vor allem durch den photoelektrischen Prozess nimmt der Massenabsorptionskoeffizient mit zunehmender Energie der einfallenden Röntgenstrahlung stetig ab. Die Kurve der Absorption gegen die Energie für ein gegebenes Element weist scharfe Diskontinuitäten auf. Diese resultieren aus charakteristischen Energien, bei denen der photoelektrische Prozess besonders effizient ist.
Scatter – Wenn Röntgenphotonen auf eine Ansammlung von Atomen auftreffen, können die Photonen mit Elektronen der Zielelemente wechselwirken, was zur Streuung der Röntgenphotonen führt, wie in Abb. 3 dargestellt. Streuung von Röntgenstrahlen von der Probe ist die Hauptquelle des Hintergrundsignals in den bei der Röntgenspektrometrie erhaltenen Spektren. Die Streuung von Röntgenstrahlen wird hauptsächlich durch äußere, schwach gehaltene Elektronen der Elemente verursacht. Wenn die Stöße elastisch sind, tritt Streuung ohne Energieverlust auf und wird als Rayleigh-Streuung bezeichnet. Wenn es unelastisch ist, verliert das Röntgenphoton Energie, um den Ausstoß eines Elektrons zu verursachen, und die Streuung ist inkohärent. Der Weg des Röntgenphotons wird abgelenkt und das Photon hat einen Energieverlust oder eine längere Wellenlänge. Das ist die Compton-Streuung.
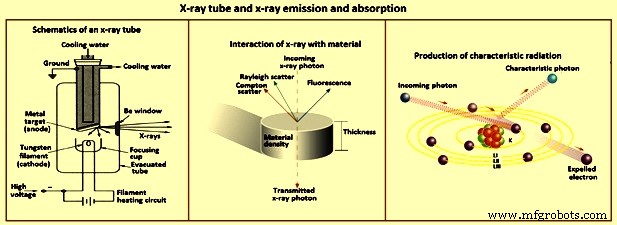
Abb. 3 Röntgenröhre und Röntgenemission und -absorption
Streuung beeinflusst die Röntgenspektrometrie auf zwei Arten. Erstens steigt die Gesamtmenge an gestreuter Strahlung mit der Ordnungszahl wegen der größeren Anzahl von Elektronen. Proben mit Matrizen mit niedriger Ordnungszahl zeigen jedoch eine größere beobachtete Streuung aufgrund der verringerten Eigenabsorption durch die Probe. Zweitens nimmt das Verhältnis der „Compton-zu-Rayleigh“-Streuintensität zu, wenn die Ordnungszahl der Probenmatrix abnimmt. Der mit der Compton-Streuung verbundene Energieverlust führt zu einer vorhersagbaren Änderung der Wellenlänge der Strahlung.
Beziehungen zwischen Elementen und Röntgenstrahlen
Die unterschiedlichen Beziehungen zwischen Elementen und Röntgenstrahlen sind in Abb. 4 dargestellt.
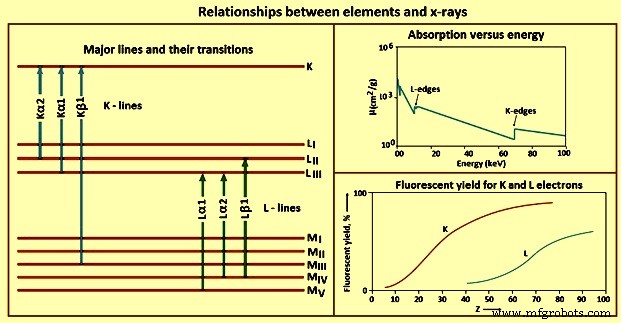
Abb. 4 Beziehungen zwischen Elementen und Röntgenstrahlen
Absorption – Röntgenphotonen können mit Orbitalelektronen von Elementen wechselwirken, um absorbiert oder gestreut zu werden. Die Beziehung zwischen Absorption und Ordnungszahl des Elements ist wichtig bei der Auswahl optimaler Betriebsbedingungen für die Röntgenspektrometrie.
Massenabsorptionskoeffizienten unterscheiden sich für ein gegebenes Element oder eine gegebene Substanz für jedes Element oder jede Substanz bei einer gegebenen Energie von Röntgenstrahlen und bei jeder Energie von Röntgenstrahlen. Aufgrund der höheren Wahrscheinlichkeit einer Wechselwirkung mit Orbitalelektronen steigt der Massenabsorptionskoeffizient mit der Ordnungszahl des Elements des Zielmaterials. Bei gegebener Ordnungszahl nimmt der Massenabsorptionskoeffizient mit der Wellenlänge der Röntgenstrahlung ab. Diese resultieren aus spezifischen Energien, die für den photoelektrischen Ausstoß von Elektronen aus den verschiedenen Orbitalen des Atoms benötigt werden und sind charakteristisch für das Element.
Absorptionskanten sind Diskontinuitäten oder kritische Punkte im Diagramm der Massenabsorption gegen die Wellenlänge oder Energie der einfallenden Röntgenstrahlung. Die Absorptionskantenenergie ist die genaue Menge, die ein Elektron aus einem Orbital eines Elements photoemittiert. Je niedriger die Hauptquantenzahl ist, desto höher ist die Energie, die benötigt wird, um ein Elektron aus dieser Schale herauszuschlagen. Die Wellenlänge eines Röntgenstrahls, der ein L-Elektron ausstoßen kann, ist länger (mit weniger Energie) als die, die benötigt wird, um ein Elektron aus der K-Schale auszustoßen. Das heißt, die K-Absorptionskantenenergie ist größer als die L-Absorptionskantenenergie für ein gegebenes Element.
Emission – Der photoelektrische Effekt ist ein Röntgenabsorptionsmechanismus, durch den instabile Zustände in den Elektronenorbitalen von Atomen erzeugt werden. Sobald die Leerstellen in den inneren Orbitalen gebildet sind, kann eine Relaxation in den stabilen Grundzustand durch die Emission von Röntgenstrahlen erfolgen, die für das angeregte Element charakteristisch sind. Die Energie der 1s Elektron ist vom Zustand der Valenzelektronen abgeschirmt, so dass die Absorptionskantenenergie und die Energie der emittierten Röntgenstrahlen im Wesentlichen unabhängig von der Oxidationsstufe und Bindung des Atoms sind.
K-Linien – Sobald der photoelektrische Effekt eine Leerstelle in der K‐Schale erzeugt, entspannt sich der angeregte Zustand, indem die Leerstelle mit einem Elektron aus einem äußeren Orbital gefüllt wird. Aufgrund quantenmechanischer Regeln, die als Auswahlregeln bezeichnet werden, sind nur bestimmte Übergänge zulässig. Die Übergänge, die den Auswahlregeln folgen, werden als erlaubte (Diagramm-)Linien bezeichnet, diejenigen, die dies nicht tun, werden als verboten bezeichnet, und diejenigen, die zum Zeitpunkt der Emission zu Atomen mit zwei oder mehr Leerstellen in inneren Orbitalen führen, werden als Satelliten (Nicht-Diagramm-Linien) bezeichnet ) Linien. Die Anzahl der K-Linien und die exakte, die für ein Element beobachtet wird, hängt teilweise von der Anzahl der gefüllten Orbitale ab.
L-Linien – Da der praktische Energiebereich für die meisten WDRFA-Röntgenspektrometer 0 keV bis 100 keV und 0 keV bis 40 keV für EDRFA-Spektrometer beträgt, muss die Verwendung anderer Emissionslinien als der K-Linien in Betracht gezogen werden. Für ein gegebenes Element werden L-Linien mit niedrigerer Röntgenenergie angeregt als K-Linien. Die Verwendung von L-Linien ist besonders wertvoll für Elemente mit Ordnungszahlen über etwa 45.
M-Linien –M-Linien finden nur begrenzt Anwendung in der routinemäßigen Röntgenspektrometrie. Die Linien werden für Elemente mit Ordnungszahlen unter etwa 57 nicht beobachtet, und wenn sie beobachtet werden, sind die Übergangsenergien niedrig. Die einzige praktische Verwendung für diese Linien ist für solche Elemente wie Thorium, Protactinium und Uran. Sie dürfen nur in diesen Fällen verwendet werden, um Interferenzen mit L-Linien anderer Elemente in der Probe zu vermeiden.
Fluoreszenzausbeute – Ein Elektron wird durch den photoelektrischen Prozess aus einem Atomorbital herausgeschleudert, mit zwei möglichen Ergebnissen, entweder Röntgenphotonenemission oder sekundärer (Auger-)Elektronenausstoß. Eines dieser Ereignisse tritt für jedes angeregte Atom auf, aber nicht für beide. Daher konkurriert die Sekundärelektronenerzeugung mit der Röntgenphotonenemission von angeregten Atomen in einer Probe. Der Anteil der angeregten Atome, der Röntgenstrahlen emittiert, wird als Fluoreszenzausbeute bezeichnet. Dieser Wert ist eine Eigenschaft des betrachteten Elements und der betrachteten Röntgenlinie. Elemente mit niedriger Ordnungszahl haben auch eine niedrige Fluoreszenzausbeute. In Verbindung mit den hohen Massenabsorptionskoeffizienten, die niederenergetische Röntgenstrahlen aufweisen, ist der Nachweis und die Bestimmung von Elementen mit niedriger Ordnungszahl durch Röntgenspektrometrie eine Herausforderung.
Inter-Element-Effekte – Bei Übergängen in der Röntgenspektrometrie hat keine Emissionslinie für eine gegebene Reihe (K, L, M) eines Elements eine Energie, die gleich oder größer als die Absorptionskante für diese Reihe ist. Ein wichtiges Ergebnis ist, dass die von einem Element emittierten Röntgenstrahlen keine Elektronen aus demselben Orbital anderer Atome dieses Elements photoemittieren können. Eine Probe, die aus einer Mischung von Elementen besteht, kann jedoch Wechselwirkungen aufweisen, die häufig als Inter-Element-Effekte bezeichnet werden. Solche Wechselwirkungen von Elementen innerhalb einer Probe erfordern häufig eine spezielle Datenanalyse.
WDRFA-Spektrometer
In den 1950er Jahren kommerziell eingeführte röntgenspektrometrische Instrumente sind als wellenlängendispersiv bekannt, was bedeutet, dass die von der Probe emittierte Strahlung unter Verwendung eines Soller-Kollimators kollimiert wird und dann auf einen Analysekristall auftrifft. Der Kristall beugt die Strahlung nach dem Braggschen Gesetz und je nach Wellenlänge bzw. Energie der Röntgenstrahlung unterschiedlich stark. Diese Winkelstreuung der Strahlung ermöglicht eine sequentielle oder gleichzeitige Detektion von Röntgenstrahlen, die von Elementen in der Probe emittiert werden.
Simultan-Instrumente enthalten normalerweise mehrere Sätze von Analysekristallen und Detektoren; einer wird für jeden gewünschten Analyten in der Probe eingestellt. Obwohl diese Instrumente teuer sind, sind sie für die routinemäßige Bestimmung von vorab ausgewählten Elementen effizient, lassen sich jedoch nicht leicht umrüsten, um andere als die bei der Installation ausgewählten Elemente zu bestimmen.
Häufiger sind sequentielle Instrumente, die ein als Goniometer bekanntes mechanisches System enthalten, das den Winkel zwischen Probe, Analysekristall und Detektor variiert. Auf diese Weise kann die gewünschte Wellenlänge der Röntgenstrahlung durch Bewegung des Goniometers ausgewählt werden. Sequentielle WDRFA-Spektrometer können zur automatischen Bestimmung vieler Elemente computergesteuert werden. Quantitative Anwendungen automatisierter WDRFA-Spektrometer sind effizient, da das Instrument so programmiert werden kann, dass es zu den richtigen Winkeln für gewünschte Bestimmungen geht. Qualitative Anwendungen sind jedoch weniger effizient, da das Spektrum langsam abgetastet werden soll.
Röntgenröhren – Verschiedene Energiequellen können verwendet werden, um die angeregten elektronischen Zustände in den Atomen von Elementen zu erzeugen, die Röntgenstrahlung erzeugen. Unter diesen sind Elektronenstrahlen, Strahlen geladener Teilchen und Röntgenstrahlung. Elektronenstrahlen werden bei solchen Techniken wie der Rasterelektronenmikroskopie (SEM) und der Elektronenmikrosondenanalyse auf die Probe gerichtet. Die Verwendung eines Elektronenstrahls erfordert jedoch ein Hochvakuum, um Energieverluste des Elektrons zu vermeiden. Die Röntgenspektrometrie wird am besten als vielseitiges Analysewerkzeug und nicht als Spezialwerkzeug eingesetzt. Viele Proben sind nicht für ein Hochvakuum geeignet oder sind Nichtleiter, die unter einem Elektronenstrahl Probleme mit der elektrischen Aufladung verursachen. Daher ist diese Energiequelle für die Röntgenspektrometrie nicht praktikabel.
Radioaktive Isotope, die Röntgenstrahlung aussenden, sind eine weitere Möglichkeit, Atome zur Aussendung von Röntgenstrahlung anzuregen. Jedoch ist der Röntgenfluss von isotopischen Quellen, der in einem Labor sicher gehandhabt werden kann, für eine praktische Anwendung zu schwach. Da diese Quellen normalerweise nur wenige schmale Röntgenlinien emittieren, werden mehrere benötigt, um viele Elemente effizient anzuregen. Die praktischste Energiequelle für die Röntgenspektrometrie ist eine Röntgenröhre (Abb. 3).
WDRFA-Spektrometer benötigen eine effiziente Hochleistungsanregung, um gut zu funktionieren. Daher ist Stabilität und Zuverlässigkeit der Röntgenröhre wichtig. Alle Komponenten befinden sich im Hochvakuum. Eine Wendel wird durch eine Wendelspannung von 6 V bis 14 V erhitzt. Die erhitzte Wendel gibt thermisch Elektronen ab. Der Elektronenfluss, der zwischen dem Filament und der Targetanode fließt, muss stark reguliert und kontrolliert werden. Dieser Elektronenfluss ist elektrischer Strom und wird normalerweise in Milliampere gemessen. Der Röhrenstrom wird oft als mA bezeichnet.
Zwischen Filament (Kathode) und Targetanode wird ein Potential von mehreren Kilovolt angelegt, das als Beschleunigungspotential für die Elektronen dient. Diese Spannung wird normalerweise in Kilovolt gemessen. Die Anode ist normalerweise Kupfer, und die Targetoberfläche ist mit hochreinen Ablagerungen solcher Elemente wie Rhodium, Silber, Chrom, Molybdän oder Wolfram plattiert. Röntgenröhren, die für die WDRFA-Spektrometrie verwendet werden, arbeiten mit 2 kW bis 3 kW. Ein Großteil dieser Leistung wird als Wärme abgeführt, und Vorkehrungen für eine Wasserkühlung der Röntgenröhre sind erforderlich. Die Stromversorgungen und die zugehörige Elektronik für diese Röntgenröhren sind groß. Die Elektronen treffen auf das Target mit einer maximalen kinetischen Energie, die dem angelegten Röhrenpotential entspricht. Wenn die kinetische Energie des Elektrons die Absorptionskantenenergie übersteigt, die dem Ausstoß eines Elektrons der inneren Umlaufbahn aus Atomen des Zielmaterials entspricht, emittiert die Röhre Röntgenstrahllinien, die für das Zielelement charakteristisch sind. Auch die Wechselwirkung der Elektronen im Strahl mit Elektronen des Zielelements führt zur Emission eines Kontinuums. Die Fläche des Kontinuums und die Wellenlänge der maximalen Intensität hängen vom Potential, Strom und der Anodenzusammensetzung ab.
Kristalle analysieren – Von der Röntgenröhre emittierte Röntgenstrahlen werden auf die Probe gerichtet. Bei den meisten Röntgenspektrometern wird die Probe in einer sogenannten Umkehroptik über der Röntgenröhre platziert. Dies erleichtert das Positionieren der Oberfläche einer Flüssigkeit unter Verwendung der Bodenfläche statt der Oberseite. Die von der Probe emittierte Röntgenstrahlung wird kollimiert und trifft auf die Oberfläche eines Analysekristalls, der die Strahlung streut. Der parallele Strahl polychromatischer Röntgenstrahlung von der Probe wird an verschiedenen Gitterebenen im Kristall gebeugt. Eine Verstärkung tritt auf, wenn die zusätzliche Strecke, die die Strahlung durch Beugung an verschiedenen Gitterebenen zurücklegen muss, gleich einem ganzzahligen Vielfachen der Wellenlänge ist. Ist dies nicht der Fall, findet destruktive Interferenz statt. Das Braggsche Gesetz erlaubt die Berechnung des Winkels, in dem eine Wellenlänge für den Analysekristall ausgewählt werden soll.
Detektoren – Detektoren und zugehörige Elektronik im WDRFA-Spektrometer erkennen Röntgenstrahlen, die vom Analysekristall gebeugt werden, und unterdrücken unerwünschte Signale wie Beugung höherer oder niedrigerer Ordnung durch den Analysekristall oder Detektorrauschen. Normalerweise werden zwei Detektoren hintereinander angeordnet. Der erste ist ein mit Gas gefüllter oder mit Gas strömender proportionaler Detektor. Diese Detektoren bestehen aus einem vom Gehäuse isolierten Draht. Dünne Polymerfenster in der Vorder- und Rückseite des Gehäuses ermöglichen den Eintritt und möglichen Austritt von Röntgenstrahlung. Zwischen Draht und Gehäuse wird ein Vorspannungspotential von einigen hundert Volt angelegt.
Obwohl viele Gase verwendet werden können, ist das typische Gas P-10, eine Mischung aus 90 % Argon (Ar) und 10 % Methan. Wenn Röntgenstrahlen in den Detektor eintreten, wird das Argon ionisiert, um viele Ar+-e-Paare zu erzeugen. Der anodische Draht sammelt die Elektronen, und die Elektronen an den kathodischen Wänden des Gehäuses neutralisieren die Ar+-Ionen. Das Ergebnis ist ein Stromimpuls für jedes Röntgenphoton, das in den Detektor eintritt. Die mit P-10 gefüllten proportionalen Detektoren sind am effizientesten zum Nachweis von Röntgenphotonen mit Energien von weniger als etwa 8 keV (Wellenlängen größer als etwa 0,15 nm). Energetischere Röntgenstrahlung neigt dazu, den proportionalen Detektor zu passieren.
Ein hinter dem Proportionalzähler häufig angeordneter zweiter Detektor ist normalerweise ein Szintillationsdetektor. Dieser Detektor besteht aus einem mit Thallium dotierten Natriumjodidkristall [NaI(Tl)], der einen blauen (410 nm) Lichtblitz aussendet, wenn er von einem Röntgenphoton getroffen wird. Der Kristall ist auf einer Photomultiplier-Röhre montiert, die die Lichtimpulse detektiert. Die Anzahl der erzeugten Lichtphotonen ist proportional zur Energie des einfallenden Röntgenphotons. Nach elektronischer Verarbeitung wird der Szintillationsstoß in einen Spannungsimpuls umgewandelt, dessen Amplitude proportional zur Röntgenphotonenenergie ist. Diese beiden Detektoren können unabhängig voneinander oder gleichzeitig betrieben werden. Im Simultanbetrieb müssen das Betriebspotential und die Ausgangsverstärkung des Detektors so eingestellt werden, dass ein Röntgenphoton einer gegebenen Energie von beiden Detektoren die gleiche Pulshöhenspannung erzeugt. Beide Detektortypen benötigen etwa 1 Mikrosekunde, um sich zwischen den Impulsen zu erholen. Einige Zählungen können bei einfallenden Photonenraten von mehr als etwa 30.000/s verloren gehen. Die Pulshöhendiskriminierung der Röntgenpulse von dem/den Detektor(en) weist Röntgenstrahlen höherer oder niedrigerer Ordnung zurück, die von dem Analysekristall gebeugt werden.
Grundlagen des Betriebs – Wenn eine Probe betrachtet und das Analytelement ausgewählt wird, ist die erste Entscheidung die Auswahl der Emissionslinie. In Abwesenheit spezifischer Interferenzen wird typischerweise die energiereichste Linie verwendet, die plausibel ist. Für Elemente mit Ordnungszahlen kleiner als etwa 75 ist dies normalerweise die K-Linie, da viele WDRFA-Spektrometer mit 100-kV-Potentialen für die Röntgenröhren arbeiten können. Wenn möglich, wird eine Röntgenröhre gewählt, die charakteristische Linien bei Energien knapp oberhalb der Absorptionskante für die zu verwendende Linie für das Analytelement emittiert. Wenn eine solche Röhre nicht verfügbar ist, muss die Anregung durch Verwendung des Kontinuums für eine verfügbare Röntgenröhre erfolgen.
Das Potential der Röntgenröhre ist auf etwa das 1,5-fache der Absorptionskantenenergie oder höher einzustellen. Der Detektor ist nach dem zu verwendenden Wellenlängenbereich auszuwählen. Der Proportionalzähler ist für Röntgenstrahlen länger als etwa 0,6 nm, der Szintillationsdetektor für Wellenlängen kürzer als etwa 0,2 nm und beide für den Überlappungsbereich von 0,2 nm bis 0,6 nm zu verwenden. Es ist ein Analysekristall auszuwählen, der die Detektion der gewünschten Wellenlänge erlaubt. Die Mehrheit der Parameterauswahl erfolgt computergesteuert.
Energiedispersive Röntgenspektrometer
Die Verwendung eines Goniometers in WDRFA-Röntgenspektrometern basiert auf der Anforderung, die von verschiedenen Elementen in einer Probe emittierten Röntgenstrahlen in Komponenten aufzulösen. Die Verwendung einer Dispersionsvorrichtung ist bei vielen Arten der Spektroskopie üblich, um diese Aufgabe zu erfüllen. Instrumente ohne die mechanischen Komponenten sind wünschenswert, wenn eine angemessene Auflösung erreicht werden kann. Die Entwicklung von Lithium-gedriftten Siliziumdetektoren und ihre Anwendung zur Röntgendetektion Mitte der 1960er Jahre führte zu einem Bereich der spektroskopischen Analyse, der als EDXRF-Spektrometrie bekannt wurde.
Röntgenröhren, die in WDRFA-Spektrometern verwendet werden, haben eine Nennleistung von 2 kW bis 3 kW und müssen mit Wasser gekühlt werden. Die in EDXRF-Spektrometern verwendeten arbeiten mit viel geringerer Leistung und sind normalerweise luftgekühlt. Typische Röhren reichen von 9 W bis 100 W. Es sind verschiedene Anodenmaterialien erhältlich, und jeder Hersteller von Röntgenspektrometern bietet spezielle Röntgenröhrenmerkmale an. Nach vielen Versuchen des Röhrendesigns bleiben die meisten jedoch beim traditionellen „Seitenfenster“-Design, obwohl es viel kleiner ist als die in WDRFA-Spektrometern verwendeten. Ein wichtiger Faktor bei der Konstruktion der Röhre und der zugehörigen Stromversorgung ist die Stabilität der Röhre und der Spannung.
Eine Alternative zur direkten Röntgenröhrenanregung ist die Verwendung einer sekundären Zielanregung. Bei diesem Modus wird mit einer Röntgenröhre ein sekundäres Target bestrahlt, dessen charakteristische Röntgenfluoreszenz wiederum zur Anregung der Röntgenemission der Probe genutzt wird. Aufgrund des erheblichen Effizienzverlusts bei Verwendung eines sekundären Targets werden Röntgenröhren mit höherer Wattleistung benötigt als für die direkte Anregung.
Die Anregung des sekundären Ziels bietet manchmal erhebliche Vorteile. Um beispielsweise die geringen Konzentrationen von Vanadium und Chrom in einer Eisenprobe zu bestimmen, können diese Elemente mit einem sekundären Eisentarget ohne Anregung des Eisens in der Probe angeregt werden. Bei direkter Röhrenerregung ist dies schwierig. Es werden mehrere Sekundärziele benötigt, um eine breite Palette von Elementen abzudecken. Die Verwendung einer sekundären Zielanregung wurde als Quelle monochromatischer Strahlung zur Anregung unterstützt. Die Bedeutung dieses Vorteils besteht darin, dass viele der Fundamentalparameter-Computerprogramme, die verwendet werden, um Intensitäten direkt aus den grundlegenden Röntgengleichungen zu berechnen, monochromatische Anregungsstrahlung benötigen.
In der Praxis nähert sich die Sekundärzielanregung nur der idealen monochromatischen Strahlung an. Die direkte Röhrenanregung mit geeigneten Primärfiltern schneidet im Vergleich zu Sekundärzieltechniken gut ab. Daher bleibt die direkte Röntgenröhrenanregung für die größte Anzahl von Anwendungen der energiedispersiven Spektrometrie (EDS) am praktischsten. The main strength of the EDS technique lies in its simultaneous multi-element analysis capabilities. Although special cases occur in which selective excitation is desirable, this frequently can be accomplished with intelligent use of an appropriate x-ray tube and filter. Any fundamental design features which limit the simultaneous multi-element capability diminish the advantage of the EDXRF spectrometer.
Since direct x-ray tube excitation is the most common method used in EDS, there are factors which govern the selection of an x-ray tube. In wavelength-dispersive techniques, several x-ray tubes are normally available for the spectrometer. These can be changed for different applications. This is not normally the case with EDS-systems, since many WDXRF spectrometer has few if any choices of primary filters. In wavelength-dispersive techniques, it is customary to attempt to excite the desired element by the characteristic emission lines of the tube anode material, but the continuum is used more efficiently in EDXRF spectrometers. The use of EDXRF spectrometers has been enhanced by computer control of tube current and voltage and selection of the primary filter. Selection and efficient use of a single x-ray tube is important in the configuration of an EDXRF system.
Characteristic lines emitted by an x-ray tube have much larger intensity at their maxima than the continuous radiation emitted. These lines are to be used for excitation whenever possible. In addition, use of a primary filter between the x-ray tube and the sample can effectively approximate monochromatic radiation impinging on the sample from these characteristic lines. EDXRF spectrometers normally offer various x-ray tube anode materials. For selecting the x-ray tube anode material, the applications most likely to be encountered are to be considered.
The principal concern is to select an anode which has characteristic lines close to, but always higher, in energy than the absorption-edge energies to be encountered. None of the characteristic lines are to create spectral interference with elements to be determined. This includes consideration of such details as the Compton scatter peak for the characteristic lines. In addition, it is difficult to perform determinations of the element of the anode material. This is especially true with samples having low concentrations of that element.
Rhodium is a favourable tube anode material for general-purpose use. The characteristic lines of this element are efficient for the excitation of elements with absorption edges to around 15 keV. The excitation efficiency for the K lines of the transition elements is low. However, the continuum can be used efficiently in this region. Rhodium also has characteristic L lines at around 2.7 keV to 3.0 keV. These are efficient for the excitation of the K lines of low atomic number elements, such as aluminum, silicon, phosphorus, and sulphur. However, in these cases, a silver anode is preferable because of the Compton scatter radiation from the rhodium lines. The characteristic lines and the continuum from the x-ray tube can be used for excitation.
Although the elements of many samples can be excited effectively using a combination of the characteristic x-ray lines from the tube anode element and the continuum, more monochromatic radiation is sometimes desired. One such situation involves enhancing the use of fundamental-parameter computations which permit quantitative determination of elements without the need for several concentration standards. A more frequent situation is the need to reduce the background in the spectrum energy range to be used in the analysis. Use of primary filters placed between the x-ray tube and the sample can be effective in these cases and are normally incorporated under computer control in commercial spectrometers.
The object is to filter the primary radiation from the x-ray tube and selectively pass the characteristic lines of the anode element. This is accomplished using a filter made of the same element as the tube anode. Since x-rays of a given line (K, L, and so on) of an element are lower in energy than the absorption edge for that element, the photoelectric component of the mass absorption coefficient is small. Such a filter does not efficiently absorb the characteristic line emitted by the x-ray tube. The higher energy x-rays from the continuum are efficient for the photoelectric process in the filter and are highly attenuated by absorption. X-rays of lower energy than the filter material absorption edge are absorbed more efficiently as the energy decreases.
The result is x-radiation striking the sample with an intensity which is largely determined by the characteristic lines of the tube anode and that approximates monochromatic radiation. Increasing the thickness of the filter decreases the total intensity, with further gain in the monochromatic approximation.
Detectors – The selective determination of elements in a mixture using x-ray spectrometry depends upon resolving into separate components the spectral lines emitted by the different elements. This process needs an energy-sorting or wavelength-dispersing device. For the WDXRF spectrometer, this is accomplished by the analyzing crystal, which needs mechanical movement to select each desired wavelength according to Bragg’s law. Optionally, several fixed-crystal channels can be used for simultaneous measurement. In contrast, EDS is based on the ability of the detector to create signals proportional to the x-ray photon energy. Hence, mechanical devices, such as analyzing crystals, are not needed.
Several types of detectors have been used, including silicon, germanium, and mercuric iodide. The solid-state, lithium-drifted silicon detector [Si(Li)] was developed and applied to x-ray detection in the 1960s. By the early 1970s, this detector was firmly established in the field of x-ray spectrometry and was applied as an x-ray detection system for SEM and x-ray spectrometry. The Si(Li) detector provides excellent resolution. It can be considered as a layered structure. Under reversed bias of around 600 V, the active region acts as an insulator with an electric-field gradient throughout its volume.
When an x-ray photon enters the active region of the detector, photo ionization occurs with an electron-hole pair created for each 3.8 eV of photon energy. Ideally, the detector is to completely collect the charge created by each photon entry and result in a response for only that energy. Some background counts appear because of energy loss in the detector. Although these are kept to a minimum by engineering, incomplete charge collection in the detector contributes to background counts. From 1 keV to 20 keV, an important region in x-ray spectrometry, silicon detectors are efficient for conversion of x-ray photon energy into charge.
Analyzer systems – The x-ray spectrum of the sample is obtained by processing the energy distribution of x-ray photons which enter the detector. One x-ray photon entering the detector causes photo-ionization and produces a charge proportional to the photon energy. Several electrical sequences are to take place before this charge can be converted to a data point in the spectrum. A detailed knowledge of the electronics is not necessary, although an understanding of their functions is important. Upon entering the Si(Li) detector, an x-ray photon is converted into an electrical charge which is coupled to a field effect transistor (FET). The FET and the electronics comprising the preamplifier produce an output proportional to the energy of the x-ray photon. Using a pulsed optical preamplifier, this output is in the form of a step signal. Since photons vary in energy and number per unit time, the output signal, due to successive photons being emitted by a multi-element sample, resembles a staircase with various step heights and time spacing. When the output reaches a determined level, the detector and the FET circuitry reset to their starting level, and the process is repeated.
The preamplifier output is coupled to a pulse processor which amplifies and shapes the signal into a form acceptable for conversion to a digital form by an analog-to-digital converter (ADC). Amplification is necessary to match the analog signal to the full-scale range of the ADC. This process involves the energy calibration of the spectrometer. Drift in the gain and/or offset (zero) of the amplification results in errors in the energy assigned to the x-ray photons producing the signal. Hence, these calibrations are to be as stable as possible, and calibration is to be routinely checked.
The energy calibration is important for qualitative identification of the elements and for precise quantitative results when using spectrum-fitting programs. The amplifier provides gain and zero controls for calibrations. Normal operation in x-ray spectrometry is to set the time on the system clock to be used to acquire the spectrum. The processing of the pulses is not instantaneous. At high count rates, the time needed can become significant. When a pulse is detected and processing initiated, the clock is ‘stopped’ until the system is ready to process a new photon. The length of time the clock is off is called dead time; the time the clock is on is called live time. Their total is real time. The system monitors live time. If the spectrometer is operated with a 50 % dead time, the real time is twice the live time.
Processing of the pulse created by a photon is to be complete before another pulse occurs. A pulse pile-up rejector circuit blocks a pulse if it is received too soon. Once activated, the pulse pile-up rejector prevents the new signal from being processed if a second x-ray enters the detector before a prior pulse is fully processed. If analysis of the prior pulse had not yet been complete, it is also to be blocked from further processing. If this blockage is not performed, pulse pile-up occurs, resulting in an artifact which appears at energies equal to the sum of the photon energy of the first and second photons to enter the detector. These are frequently called sum peaks.
Despite pulse pile-up rejection circuitry, sum peaks are observed for intense peaks in the spectrum. This is the result of two photons entering the detector simultaneously or within a time difference faster than the fast discriminator can act. Sum peaks can be observed at twice the energy of an intense peak and / or at the sum of the energies of two intense peaks in the spectrum. Sum peaks decrease rapidly in intensity with count rate. The importance of electronic pulse-processing components to system performance is easily overlooked in EDS. However, stability, linearity, and proper calibration of these components are important for the use of the spectrometer.
EDXRF spectrometers require a dedicated computer system for data acquisition. Early spectrometers were heavy, unwieldy units which used hard-wired multichannel analyzers which could acquire data, but could do little to process it. Current spectrometer and data systems based on microprocessor technology are available as table-top units.
Fundamentals of operation – The simultaneous multi-element capability of EDS complicates the selection of optimum conditions because of the factors to be considered for each element. The compromises in spectroscopy are to be made, but the initial selection of instrument operating conditions can follow a logical sequence of decisions.
Qualitative analysis needs similar procedures, normally with less stringent requirements. Once a sample is received for analysis and the elements to be determined by x-ray spectrometry are identified, the next decision is to ascertain which x-ray lines are to be used for the determinations. As a general rule, K lines are used upto a K absorption-edge energy a few keV below the characteristic line of the x-ray tube anode element. The continuum can be used for excitation if the voltage to the x-ray tube is set sufficiently high to place the continuum maximum at energy higher than the absorption edge and if a back-ground filter is used. In these cases, K absorption-edge energies can be used upto around 66 % of the maximum operating kV of the x-ray tube. However, the observed peaks lie on a continuum background and reduce the signal-to-noise ratio.
For a 50-kV x-ray tube, absorption edges as high as 30 keV can be used if the element is present in sufficient concentration. For a 30-kV rhodium or silver tube, one is restricted essentially to excitation by the characteristic tube lines. This is of no great concern unless there is a special interest in the elements between atomic numbers 41 and 50 (niobium to tin). Elements above atomic number 50 (40 for a 30-kV system) are normally to be determined using the L lines of their x-ray spectra.
To excite all L lines, the incident x-ray photon energy is to exceed the LI absorption edge. For practical use, the energy of the L lines is to be higher than around l keV. For the L line spectra, this needs atomic numbers higher than 30. At such low x-ray energies, absorption of the x-rays and low fluorescent yield in the L emission in this region needs high concentration of the element to be determined and excellent sample preparation. Overlap of the K lines of the low atomic number elements in this region also causes difficulty. For example, the K lines of phosphorus overlap with the L lines of zirconium and the M lines of iridium at around 2 keV. These problems are to be considered, but are to a large degree solved by careful use of processing software.
Once the x-ray spectral lines are selected for determination of the elements, the next step is to decide whether all analyte elements in the sample can be determined with one instrumental setting. Although the multi-element capability of EDS is useful, all elements in every sample cannot be determined with a single set of instrument parameters. Some applications need more than one condition, such as a mixture of low atomic number elements and transition elements. The transition elements are best determined by excitation using the K lines of rhodium or silver and the low atomic number elements with the L lines or a properly adjusted continuum using a background filter. Computer control of instrument parameters facilitates changing the conditions. Whether automatic or manual control is used, all samples are to be analyzed under one set of conditions, then analyzed again using the alternate set. This is preferred over changing conditions between samples.
X-ray tube operating voltage affects the efficiency of excitation of each element in the spectrum and the integrated x-ray photon flux from the tube. The tube current affects the flux only. Hence, once the operating kV has been set, the tube current typically is adjusted until the system is processing counts efficiently. System dead time is to be maintained below, but near, 50 %. The voltage and current settings for the x-ray tube have a sensitive effect on the rate of information acquisition and count distribution among the respective spectral peaks for a given type of sample.
Selection of primary tube filter thickness is important. If the filter is changed, the tube current, and sometimes the voltage, frequently needs resetting since the filter alters the intensity distribution of the x-rays striking the sample. When characteristic tube lines are used for excitation, the filter is normally made from the tube anode element. The intensity of the transmitted x-rays decrease exponentially with increasing filter thickness. It is common to have two or three primary filters made from the tube anode element in the filter holder. The selection is to reflect optimum count rate corresponding with reasonable current and voltage settings. Thicker filters attenuate lower energy radiation more effectively and reduce the excitation efficiency for the element with low absorption coefficients.
The remaining decision is the choice of atmosphere in the sample chamber. If x-rays below around 5 keV are to be implemented, use of a vacuum can be advantageous. Intensity can increase sufficiently to reduce significantly the counting time needed to achieve an adequate number of counts. If the concentration of elements yielding these x-rays is sufficiently high, the vacuum may not be needed. Because of the extra precautions needed in sample criteria and handling, a vacuum path is not to be used unless significant benefit is realized. Similar reasoning applies to the helium atmosphere.
These guidelines are useful for initial selection of operating conditions. The instrumental parameters are interactive, and a change in one parameter needs adjustment of another. For example, selection of a thicker primary filter or a decrease in the tube voltage needs an increase in the tube current.
Sample preparation
The care taken to determine the best method of sample preparation for a given material and careful adherence to that method frequently determine the quality of results obtained. Sample preparation is the single most important step in an analysis, yet it is frequently given the least attention. In most cases, the stability and overall reproducibility of x-ray instrumentation are the least significant factor affecting the precision of analytical measurements. Frequently, the precision of analytical results expected from x-ray spectrometric determinations is expressed in terms of the theoretical statistics of measurement of x-ray intensities.
When replicate samples are prepared and actual standard deviations measured, deviations are found to be larger than those predicted by counting statistics. If precision is poor, any one analytical result can also be poor, since it can differ substantially from the ‘true’ value. The variety of sample types which can be analyzed using x-ray spectrometry necessitates different sample preparation techniques.
Samples are frequently classified as infinitely thick or infinitely thin based on measurement of the attenuation of x-rays. Samples are considered to be infinitely thick if further increase in the thickness yields no increase in observed x-ray intensity. The critical value for infinite thickness depends on the energy of the emitted x-radiation and the mass absorption coefficient of the sample matrix for those x-rays. For pure iron, the critical thickness is around 40 m for iron x-rays.
Although infinitely thin samples afford several advantages, it is rarely feasible to prepare them from routine samples. Many samples fall between these two cases and need extreme care in preparation. In addition to preparation of the sample, precise positioning of the sample in the spectrometer is critical for quantitative determinations.
Solid samples – These are defined as single bulk materials, as opposed to powders, filings, or turnings. Solid samples can frequently be machined to the shape and dimensions of the sample holder. The processing is not to contaminate the sample surface to be used for analysis. In other cases, small parts and pieces are to be analyzed as received. The reproducible positioning of these samples in the spectrometer is critical. It is frequently useful to fashion a wax mould of the part which fits into the sample holder. Using the mould as a positioning aid, other identical samples can be reproducibly placed in the spectrometer.
Samples taken from unfinished bulk material frequently needs surface preparation prior to quantitative analysis. Surface finishing can be done using a polishing wheel, steel wool, or belt grinder, with subsequent polishing using increasingly fine abrasives. Surface roughness less than 100 micrometers is normally sufficient for x-ray energies above around 5 keV, but surface roughness of less than 20 micrometers to 40 micrometers is needed for energies down to around 2 keV. Several precautions are necessary. Alloys of soft metals can smear on the surface as the sample is polished, resulting in a surface coating of the soft metal which yields high x-ray intensities for that element and subsequently high analytical results.
Polishing grooves on the surface of the sample can seriously affect the measured intensity of low-energy x-rays. This can be examined by repetitive measurement of the intensity of a sample after 45 degrees or 90 degrees rotation. Use of a sample spinner reduces this effect. If a sample spinner is not available, the sample is to be placed in the spectrometer such that the incident x-radiation is parallel to the polishing direction.
Powders and briquettes – Powder samples can be received as powders or prepared from pulverized bulk material too inhomogeneous for direct analysis. Typical bulk samples pulverized before analysis are ores, and refractory materials. Powders can be analyzed using the spectrometer, pressed into pellets or briquettes, or fused with a flux, such as lithium tetra borate. The fused product can be reground and pressed or cast as a disk. For precise quantitative determinations, loose powders are rarely acceptable, especially when low-energy x-rays are used. Pressed briquettes are more reliable. However, experience indicates that the best compromise is reground and pressed fusion products. This technique eliminates several problems associated with particle-size effects.
Particle-size effects result from the absorption of the incident and emitted x-rays within an individual particle. If the mass absorption coefficient of the sample matrix is high for the x-radiation used, particles even a few microns in diameter can considerably affect attenuation of the radiation within each particle. If the sample consists of particles of various sizes, or the particle size varies between samples, the resulting x-ray intensities can be difficult to interpret. This problem is compounded by the tendency of a material composed of a mixture of particle sizes to segregate when packed.
Determination of elements using low-energy x-radiation can lead to errors from particle-size effects of as much as 50 %. If the needed speed of analysis prohibits use of fusion techniques, direct determination from packed powders can be considered. The sample is to be ground, if possible, to a particle size below the critical value. The grinding time needed frequently can be ascertained by measuring the intensity from a reference sample at increasing grinding times until no further increase is observed. The lowest energy x-ray to be used in analysis is to be selected for this test.
Briquettes or pressed powders yield better precision than packed powder samples and are relatively simple and economical to prepare. In several cases, only a hydraulic press and a suitable die are needed. In the simplest case, the die diameter is to be the same as the sample holder so that the pressed briquettes fit directly into the holder. The amount of pressure needed to press a briquette which yields maximum intensity depends on the sample matrix, the energy of the x-ray to be used, and the initial particle size of the sample. Hence, prior grinding of the sample to a particle size which is less than 100 micrometers is advisable.
A series of briquettes are to be prepared from a homogeneous powder using increasing pressure. The measured intensity of the x-ray lines to be used in the analysis is plotted versus the briquetting pressure. The measured intensity is to approach a fixed value, perhaps asymptotically. Pressures of 138 MPa to 276 MPa may be needed. For materials which do not cohere to form stable briquettes, a binding agent is needed. Acceptable binding agents include powdered cellulose, detergent powders, starch, stearic acid, boric acid, lithium carbonate, polyvinyl alcohol, and commercial binders.
Briquettes which are not mechanically stable can be improved by pressing them into backing of pre-pressed binder, such as boric acid, or by the use of a die which presses a cup from a binding agent. The sample powder can then be pressed into a briquette supported by the cup. Improved results are frequently achieved if around 0.1 mm to 0.5 mm is removed from the surface of the briquette prior to the measurement.
Fusion of materials – Fusion of materials with a flux can be performed for several reasons. Some refractory materials cannot be dissolved, ground into fine powders, or converted into a suitable homogeneous form for x-ray spectrometric analysis. Other samples can have compositions which lead to severe inter-element effects, and dilution in the flux reduces these. The fused product, cast into a glass button, provides a stable, homogeneous sample well suited for x-ray measurements. The disadvantages of fusion techniques are the time and material costs involved as well as the dilution of the elements which can result in a reduction in x-ray intensity. However, when other methods of sample preparation fail, fusion frequently provides the needed results.
Low-temperature fusions can be carried out using potassium pyro-sulphate. More common are the glass-forming fusions with lithium borate, lithium tetra-borate, or sodium tetra-borate. Flux-to-sample ratios range from 1:1 to 10:1. The lithium fluxes have lower mass absorption coefficients and hence less effect on the intensity of the low-energy x-rays. An immense variety of flux-additive recipes are reported for various sample types. Lithium carbonate can be added to render acidic samples more soluble in the flux. Lithium fluoride has the same effect on basic samples. Lithium carbonate also reduces the fusion temperature. Oxidants, such as sodium nitrate and potassium chlorate, can be added to sulphides and other mixtures to prevent loss of these elements.
Filters and ion-exchange resins – Various filters, ion-exchange resin beads, and ion-exchange resin-impregnated filter papers have become important sampling substrates for samples for x-ray spectrometric analysis. Filter materials can be composed of filter paper, membrane filters, glass fiber filters, and so on. Filters are used in a variety of applications. One widely used application is in the collection of aerosol samples from the atmosphere. Loadings of several milligrams of sample on the filter can correspond to sampling several hundred cubic meters of atmosphere. Such sampling can be performed in any environment. Many elements can be determined directly on these filters by x-ray spectrometric analysis. Particulate samples collected in this way present problems, stemming primarily from particle-size effects, which are reduced in part by the need to collect two particle-size regions using dichotomous samplers. With these units, particles are separated into those smaller and those larger than around 2 micrometers in diameter.
The smaller particles tend to represent man-made materials; the larger ones are of natural origin. The smaller particles show fewer particle-size effects, and an x-ray spectrometric determination of even low atomic number elements, such as sulphur, is possible. Glass fiber filters are frequently used for this purpose. Filters can also be used for non-aerosol atmospheric components, such as reactive gases. Filter materials can be impregnated with a reagent reactive to the gas which traps it chemically. Sampling is accomplished by conveying atmospheric gases through a treated filter under carefully controlled conditions. An example is a damp filter treated with ferric ion solution used to trap hydrogen sulphide. The excess iron can be rinsed from the filter, but the precipitated ferrous sulphide remains. The sulphur can be determined directly or indirectly by measuring the iron x-radiation. The key to determining atmospheric components is the development of suitable standards.
Filters can be used to determine solution components in ways parallel to those described for atmospheric components. Particulate materials can be filtered directly from solution. For example, particulate materials in environmental water samples are defined as that which is filtered using a 0.45 micrometer pore diameter membrane filter. Hence, filtration of particles from water can be accomplished using such filters, and direct x-ray spectrometric analysis performed. Application of filter sampling to dissolved elements in water is becoming more common. The principle is similar to the reactive reagent-impregnated filter application to atmospheric gases. In some cases, the filter can be impregnated with ion-exchange resins which trap ions as the solution passes through the filter.
Procedures using ion-exchange resin-impregnated filters are to be carefully checked, since several passes of the solution can be needed, and distribution of the ions across the paper thickness is seldom uniform. However, for solutions, a reaction can be performed prior to filtration. For example, many ions can be precipitated quantitatively from aqueous solution, even at parts per billion concentration levels. The precipitates can be collected using 0.45 micrometers pore diameter membrane filters, which are then mounted between two Mylar sheets retained by ring clips on a standard plastic sample cup. Simultaneous multi-element determinations are then performed using XRF spectrometer.
Detection limits on the filters of as low as a few tenths of a microgram are common. If 100 g of sample solution is used, this corresponds to the detection limits of a few parts per billion in the sample. Standards are easily prepared as aqueous solutions. ‘Standard reference materials’ (SRM) for environmental waters and industrial effluent water are available.
Thin-film samples – Thin-film samples are ideal for x-ray spectrometric analysis. The x-ray intensity of an infinitely thin sample is proportional to the mass of the element on the film, and the spectral intensities are free of inter-element and mass absorption coefficient effects. However, in practice, perfect thin-film samples are rarely encountered. Powder samples of sufficiently small and homogeneous particle size can be distributed on an adhesive surface, such as cellophane tape, or placed between two drum-tight layers of Mylar film mounted on a sample cup.
More important thin-film types are platings and coatings on various substrates. Analysis of these sample types is increasingly important for the electronics industry. Of particular concern are measurements of film thickness and composition. Several techniques can be used, including the substrate intensity attenuation method, the coating intensity method, various intensity ratio methods, and the variable takeoff angle method. The last method is not practical in most commercial spectrometers. To be infinitely thin to most x-rays used in x-ray spectrometric analyses, the samples are to be 10 micrometers to 200 micrometers thick.
Liquids – Liquids can also be analyzed using x-ray spectrometry. The design of x-ray spectrometric instrumentation using inverted optics, in which the sample is above the x-ray source and detector, facilitates the use of liquid samples. This convenient geometry demands caution in the preparation of liquid samples to avoid damaging the source or detector by such accidents as spills and leaking sample cups.
Quantitative standards are easily prepared for liquid samples. However, since solvents are normally composed of low atomic number elements, the Rayleigh and Compton scatter intensity is high, which increases background and leads to high limits of detection. These problems can be minimized by use of suitable primary tube filters, which reduce the scattered x-radiation in the analytically useful region.
Care is to be taken with liquids containing suspended solids. If the suspension settles during the measurement time, the x-ray intensity of the contents of the sediment is enhanced. The x-ray intensity from solution components or homogeneous suspension can decrease as a result of sediment absorption, which leads to erroneous results. This possibility is tested by brief, repetitive measurements, beginning immediately after a sample is prepared. Any observed increase or decrease in intensity with time indicates segregation in the sample. In these cases, an additive which stabilizes the suspension can be used, or the suspended content can be collected on a filter for analysis.
Special sample types – Applications of x-ray spectrometric analysis do not always provide convenient samples which can fit one of the above categories. Non-destructive analyses are occasionally needed on production products which are not 32 mm diameter circles of infinite thickness. Examples include computer disks, machined parts, and long, coated strips or wire. In these cases, a sample compartment which accommodates the samples can frequently be designed. With the development of the mercuric iodide detector, which can provide adequate resolution for many analyses without a liquid nitrogen dewar, special analytical systems for on-line and non-destructive analysis of large samples can become increasingly feasible.
Herstellungsprozess