


Entschlüsselung der SARS-CoV-2-Genome – Herkunft
Die anhaltende COVID-19-Pandemie ist eine globale Bedrohung für die öffentlichen Gesundheitssysteme und hat die Weltwirtschaft hart getroffen. Der Übeltäter dieser Pandemie, SARS-CoV-2, ist nicht die typische Grippe. Dieses Virus befällt sowohl die oberen als auch die unteren Atemwege, stört den Kernprozess des Lebens, die Atmung und ist daher tödlich. Bis zum 6. April 2020 hat Worldometer 1.337.166 Fälle mit 74.176 Todesfällen weltweit gemeldet.
Die Untersuchung von SARS-CoV-2 auf Genomebene wird Einblicke in das Verständnis der Ursprünge dieses Virus geben. Es wird Wissenschaftlern auch dabei helfen, Diagnosewerkzeuge zu entwickeln, um diesen unsichtbaren Krankheitserreger zu erkennen und die Erfindung von Therapeutika zu erleichtern, um den Verlust von Menschenleben zu minimieren.
Das SARS-CoV-2-Genom verstehen
Ein Virus ist ein infektiöses Agens, das einen lebenden Wirt benötigt, um zu gedeihen und sich zu vermehren. Außerdem ist SARS-COV-2 ein einzelsträngiges RNA-Virus mit einem Genom von fast 30 kb Nukleotidbasen mit 12 mutmaßlichen offenen Leserahmen. Kurz nach Ausbruch der Epidemie im Dezember 2019 sequenzierten chinesische Wissenschaftler das SARS-CoV-2-Genom. Verschiedene wissenschaftliche Gruppen haben in den letzten Wochen komplette Genomsequenzen von SARS-CoV-2 veröffentlicht. Diese sind öffentlich in der Genbank und der Coronavirus-Datenbank verfügbar.
Ursprung des SARS-CoV-2-Virus
Bei solchen Ausbrüchen können nichtwissenschaftliche Verschwörungstheorien zu unnötigen Vorurteilen gegenüber Ländern, Gemeinschaften und Kulturen führen. SARS-CoV-2 ist keine Ausnahme, und die Situation wird durch die heutigen wachsenden Social-Media-Plattformen noch verschärft. Es obliegt uns, diesen unsichtbaren Feind durch eine rationale wissenschaftliche Linse zu betrachten. Basierend auf Genomanalysen ist SARS-CoV-2 ein Virus, das sich natürlich entwickelt und ist kein synthetischer Laborstamm 1,2 . Wissenschaftler haben die vollständigen Genome von mehr als 100 SARS-CoV-2-Stämmen aus verschiedenen Regionen der Welt sequenziert. Es stellt sich heraus, dass diese Stämme auf Nukleotidebene zu mehr als 99,5 % identisch sind. Dies deutet darauf hin, dass die Stämme in verschiedenen Regionen nicht viel mutierten, angeblich weil das Virus bereits eine hohe Infektionsrate und Virulenz aufweist.
In der jüngsten Vergangenheit haben zwei weitere Coronaviren weltweite Aufmerksamkeit erregt. Dies waren das SARS-CoV, China, 2002, und das MERS-CoV, Saudi-Arabien, 2012. Es wurde gezeigt, dass diese beiden früheren Viren von Fledermäusen stammen. Basierend auf diesem historischen Wissen sequenzierten Wissenschaftler das Coronavirus von Fledermäusen und zeigten, dass Bat CoV (RaTG13 ) war zu 96,2 % identisch mit SARS-COV-2, was den zoonotischen Ursprung des letzteren bestätigt. 2 Das Coronavirus verwendet oft einen Zwischenträger, bevor es den Menschen befällt. Interessanterweise veranlassten um den Oktober 2019 Berichte über tote malaiische Schuppentiere mit Lungen- und Lungenschaumfibrose-Symptomen im Guangdong Wildlife Rescue Center in China die Wissenschaftler, ihr Metagenom zu isolieren. Tatsächlich enthielten die Metagenomdaten der toten Schuppentiere das Coronavirus! 3
Interessanterweise ist SARS-CoV-2 auf der gesamten Genomebene zu fast 91% identisch mit dem malaiischen Schuppentier-CoV, was darauf hindeutet, dass Schuppentiere ein Zwischenwirt sein könnten.
Was sind Schuppentiere? Sie sind ameisenfressende Säugetiere, die in Asien sowohl für die traditionelle chinesische Medizin als auch für ihr Fleisch, das viele als Delikatesse bezeichnen, sehr gefragt sind. Sie sind auch heute die am häufigsten gehandelten Säugetiere im illegalen Handel mit Wildtieren.
SARS-CoV-2 unterscheidet sich von anderen bekannten Coronaviren mit einer Sequenzidentität von 88% oder weniger. Basierend auf phylogenetischen Analysen ist SARS-CoV-2, das bei Menschen, Fledermäusen (RaTG13) und malaiischen Schuppentieren beobachtet wurde, eine neue Klasse von Beta-Coronaviren. Fast 35 verschiedene Arten von Coronavirus-Stämmen aus verschiedenen Teilen der Welt und von verschiedenen Organismen wurden auf der gesamten Genomebene analysiert. SARS-CoV-2, unten blau dargestellt, ist eine neue Klasse von Beta-Coronaviren (Abbildung 1).
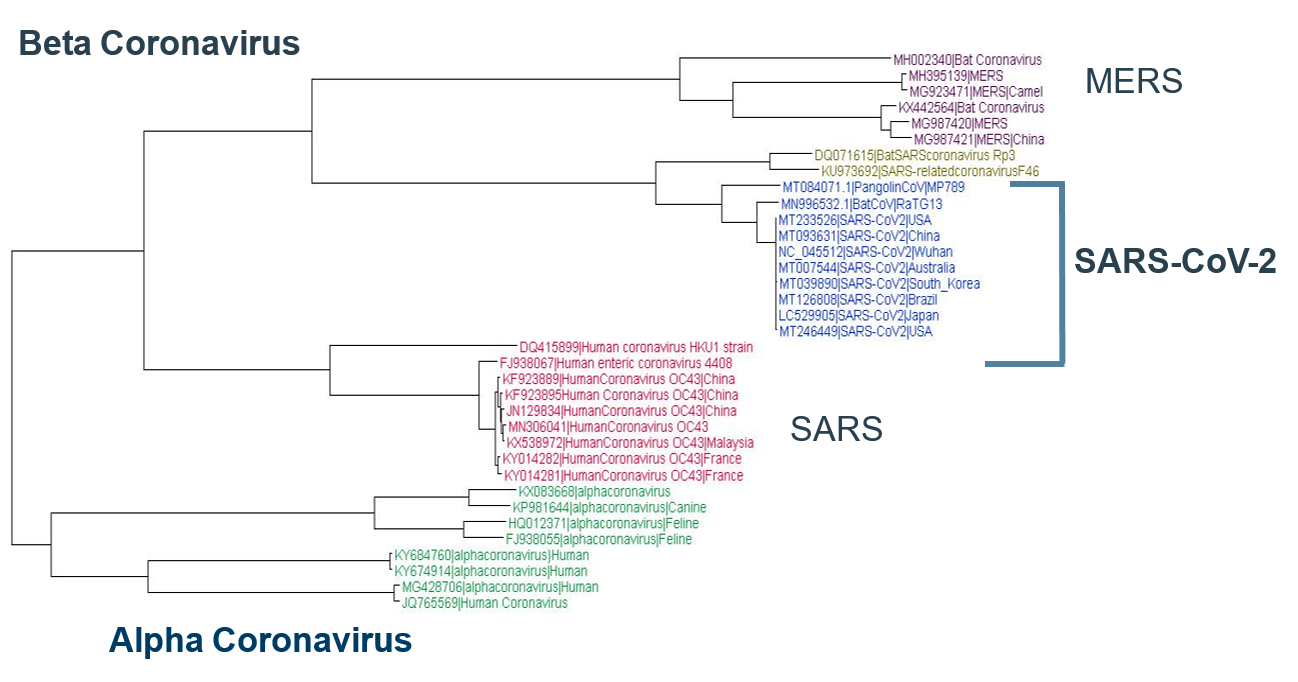
Wie dringt das Coronavirus in den Gastgeber ein?
Eine wichtige Rolle scheint dabei eines der Proteine des Coronavirus zu spielen, das als Spike-Protein bezeichnet wird. Das Spike-Protein ist eine multifunktionale molekulare Maschine, die aus zwei Hauptuntereinheiten, S1 und S2, besteht. Das Spike-Protein bindet zuerst über seine S1-Untereinheit an einen Rezeptor auf der Wirtszelloberfläche und verschmilzt dann Virus- und Wirtsmembranen über seine S2-Untereinheit. Die Domäne in S1 verschiedener Coronaviren erkennt eine Vielzahl von Wirtsrezeptoren, was zu einer viralen Anheftung führt. Die Rezeptorbindungsdomäne (RBD), die aus 193 Aminosäuren besteht, bindet und verbindet sich mit der Wirtszelle. Der Rezeptor für SARS-CoV-2 beim Menschen ist das Angiotensin Converting Enzyme 2 (ACE2). ACE2 ist an der äußeren Oberfläche von Zellmembranen in Lunge, Arterien, Herz, Niere und Darm befestigt. ACE2 senkt den Blutdruck, indem es die Spaltung von Angiotensin II, einem vasokonstriktorischen Peptid, in Angiotensin1-7, einen Vasodilatator, katalysiert. Leider scheint ACE2 auch ein beliebter Einstiegspunkt für Coronaviren zu sein.
Die Sequenzen von Pangolin CoV und SARS-CoV-2 sind in der RBD-Region hoch konserviert, was darauf hindeutet, dass das pathogene Potenzial des Virus zwischen Pangolin CoV und SARS-CoV-2 sehr ähnlich ist. Die Schlüsselaminosäurereste, die die Bindung bestimmen, sind bei Pangolin CoV und SARS-CoV-2 im Sequenz-Alignment identisch (in Abbildung 2a mit blauen Kästchen markiert) und den Schlüsselaminosäuren (LFQSNY) oberhalb der Cartoons in Abbildung 2b . Interessanterweise unterscheidet sich die SARS-CoV-2-RBD der Fledermaus in 17 Aminosäureresten, darunter fünf kritische Reste für die Bindung 3 . Basierend auf der Analyse der Sequenzdaten kann man spekulieren, dass das Fledermaus-SARS-CoV-2 möglicherweise nicht die Schlüsselreste besitzt, um an das ACE2-Protein der Wirtszelle zu binden, um eine Infektion auszulösen. Dies erfordert Experimente, um dies zu bestätigen.
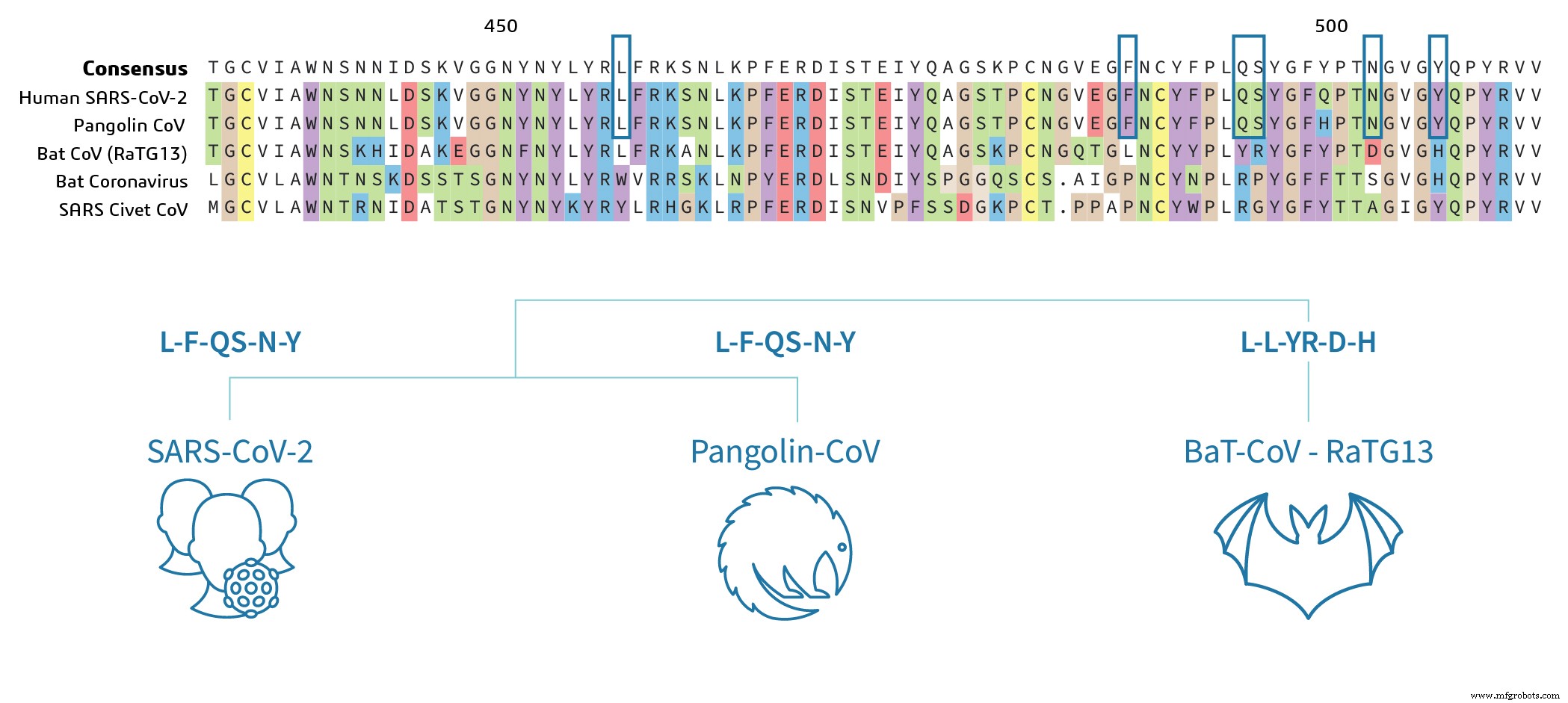
Wie bereits erwähnt, enthält das Spike-Protein zwei funktionelle Domänen:eine Rezeptorbindungsdomäne und eine zweite Domäne, die Sequenzen enthält, die die Fusion der Virus- und Zellmembranen vermitteln. Das Spike-Glykoprotein muss von Zellproteasen gespalten werden, um das Freilegen der Fusionssequenzen zu ermöglichen und wird daher für den Zelleintritt benötigt. Der Vergleich der S1/S2-Schnittstellensequenz von Pangolin-CoV und Fledermaus-SARS-CoV-2 zeigt eine Insertion des Furin-Erkennungsmotivs. Dies weist auf einen unterschiedlichen Mechanismus für den Eintritt des viralen Genoms in das Zytoplasma des Wirts zur Replikation hin, wie in Abbildung 3 gezeigt.
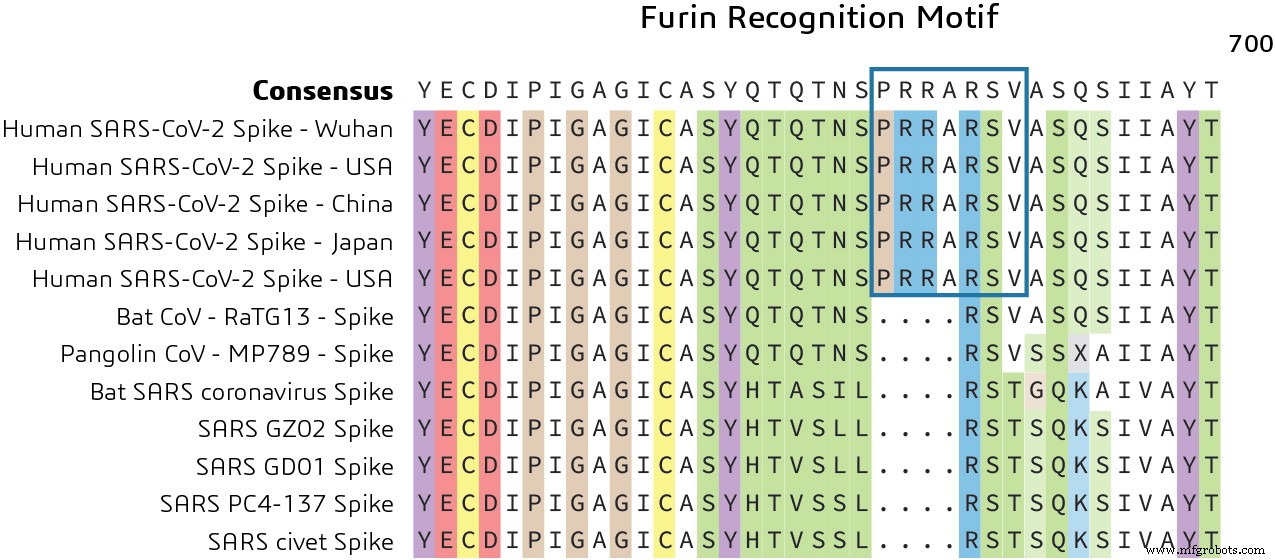
Welche Rolle spielt das Furinerkennungsmotiv? Beim Menschen wird das Furin-Erkennungsmotiv (PRRARSV) vom FURIN-Protein erkannt, einem Mitglied der S8-Familie von Subtilisin-ähnlichen Peptidasen, das dabei hilft, Abschnitte des Proteins zu entfernen, um ihre Konformation von einem inaktiven in einen aktiven Zustand zu ändern.
Es wurde vermutet, dass der Erwerb dieser Furin-Spaltungsstelle ein „Funktionsgewinn“ sein könnte, der es einem Fledermaus-CoV ermöglichte, in den Menschen zu springen und seine aktuelle epidemische Ausbreitung zu beginnen. Dies könnte ein potenzieller Weg für die Erforschung neuer Medikamente sein, die auf die Blockierung dieses Motivs abzielen, um die Replikation des Virus im Wirt zu verhindern.
So zeigt eine sorgfältige Untersuchung des Spike-Proteins in SARS-CoV-2 das optimierte RBD, ein Furin-Erkennungsmotiv, wie einige MERS-Coronaviren, und seine Fähigkeit, stark an das ACE2-Protein zu binden. Dies deutet auf einen natürlichen Selektionsprozess im Spiel hin. Es hat sich gezeigt, dass natürliche Rekombinationsereignisse bei Viren, die einen Wirt koinfizieren, ihr Wirtsspektrum verbessern und gleichzeitig die Virulenz und Virusadaptation erhöhen. SARS-CoV-2-Genomdaten mit dem Rückgrat der Fledermaus (RaTG13) und Schuppentier-CoV weisen erneut darauf hin, dass es sich um ein Virus handelt, das durch natürliche Rekombination erzeugt wird .
Was ist der Sofortspender von SARS-CoV-2 für den Menschen?
Die SARS-CoV-2-Sequenz enthält eine Mischung aus Fledermaus-SARS-CoV (RaTG13) sowie Regionen von konserviertem Pangolin-CoV, die nur während der Rekombination dieser viralen Genome auftreten können. Auch ein Funktionsgewinn, wie er bei dem Furinerkennungsmotiv zu sehen ist, beinhaltet eine weitere Virusrekombination. Damit eine Rekombination stattfinden kann, ist es nur logisch, dass es einen natürlichen Wirt gibt, der diese viralen Genome beherbergt. Ist es ein anderes Schuppentier? Oder ein anderes wildes Tier auf dem Wuhan-Meeresfrüchtemarkt? Dies ist noch unbekannt. Das Verständnis des Ursprungs könnte dazu beitragen, zukünftige Ausbrüche von Virusstämmen und globale Pandemien zu verhindern.
Für weitere Informationen kontaktieren Sie uns bitte:https://www.3dsbiovia.com/about/contact/.
Biologie
- Der 555 IC
- Die quadratische Formel
- Dekodierung von ‚Industrie 4.0‘
- Ein Supply-Chain-Ansatz zur Lösung der Coronavirus-Herausforderung
- Wird die Coronavirus-Epidemie als Weckruf für globale Lieferketten dienen?
- Das Coronavirus erschüttert traditionelle Lieferketten
- Sechs Lieferkettenstrategien für Öl und Gas in Zeiten des Coronavirus
- Das Coronavirus könnte das Ende schlechter Versanddaten begünstigen
- Wie Data Science bei der Bekämpfung des Coronavirus-Ausbruchs half
- 5 Ws des SARS-CoV-2 RapidPlex Sensors