


Pullulan-basierte Nanopartikel-HSA-Komplexbildung und Wirkstofffreisetzung beeinflusst durch Oberflächenladung
Zusammenfassung
Die Nanomaterialzusammensetzung von Nanopartikeln und deren Proteinadsorption im Blut ist von großer Bedeutung für das Design von wirkstoffbeladenen Nanopartikeln. Um die Wechselwirkung zwischen den verschiedenen Oberflächenkomponenten von Nanopartikeln (NPs) und Proteinen zu untersuchen, synthetisierten wir drei Arten von Pullulan-NP-Polymeren:cholesterisches hydrophob (CH) modifiziertes Pullulan (CHP), CH-modifiziertes animiertes Pullulan (CHAP) und CH-modifiziertes carboxyliertes Pullulan (CHSP). Pullulan-NPs wurden nach dem Dialyseverfahren hergestellt. Die dynamische Lichtstreuung wurde verwendet, um die Ladung und Größe der drei NPs zu bestimmen. Die Größe der NPs wurde durch die Anzahl der Ladungsgruppen verändert, wenn Polymere den gleichen Grad an Cholesterinsubstitution aufweisen. Die Zetapotentiale betrugen + 12,9, − 15,4 und − 0,698 mV für CHAP, CHSP bzw. CHP, und die Abmessungen betrugen 116,9, 156,9 bzw. 73,1 nm. Mit isothermer Titrationskalorimetrie wurden die thermodynamischen Veränderungen von NPs mit unterschiedlicher Oberflächenladung bestimmt und der Einfluss von Humanserumalbumin (HSA) auf die Titration untersucht. Die Änderungen von Enthalpie und Entropie zeigten eine Wechselwirkung zwischen NPs und HSA; die Bindungskonstante (K b ) für CHSP, CHP und CHAP betrug 1,41, 27,7 und 412 × 10 4 M −1 , mit positiver Ladung für CHAP-HSA, ungeladen für CHP-HSA und negativer Ladung für CHSP-HSA-Komplex. Fluoreszenz- und Circulardichroismus-Spektroskopie wurden verwendet, um die Proteinstrukturänderung nach der Komplexierung zwischen NPs und HSA zu bestimmen. Die NP- und HSA-Komplexierung ist ein komplizierter Prozess, der aus der Reduzierung des Protein-α-helikalen Gehalts und der Peptidkettenverlängerung besteht; CHP-NPs wiesen die größte Verringerung des HSA-α-helikalen Gehalts auf. Die Wirkstofffreisetzungsraten aller Verbindungen von NP und HSA waren nach 48 h signifikant niedriger als die von freien Wirkstoffen und wirkstoffbeladenen NPs. Die höchsten und niedrigsten Raten wurden bei CHSP-HSA bzw. CHP-HSA beobachtet. Die Wirkstofffreisetzung wurde maßgeblich durch die Adsorption von HSA an NPs beeinflusst, und die Größe und Oberflächenladung der NPs spielte dabei eine wichtige Rolle.
Hintergrund
Nano-Wirkstoff-Verabreichungssysteme, wie Nanopartikel (NPs), die mit kleinmolekularen Antitumor-Wirkstoffen beladen sind, haben anhaltende und kontrollierte Wirkstofffreisetzungseigenschaften sowie eine zielgerichtete Wirkung. Die gezielte Therapie mit NPs ist zu einem Schwerpunkt bei der Behandlung von Tumoren geworden, da sie die Nebenwirkungen von Medikamenten deutlich reduzieren und die Wirksamkeit von Medikamenten verbessern können [1,2,3].
Wirkstoffbeladene NPs müssen drei Wege durchlaufen, um den Zielort zu erreichen, d. h. Blutzirkulation, Gewebe-Zell-Weg und intrazelluläre Bewegung [4,5,6]. Sie müssen auch die Gefäßbarriere überwinden, um das Zielgewebe zu erreichen, und dann die Zellmembranbarriere, um die Zielzelle zu erreichen [7, 8]. Proteinadsorption und -austausch sind an allen Reaktionswegen aller NPs beteiligt. Schließlich erreichen proteinadsorbierte NPs die Zielzellen und setzen die Medikamente frei [1, 9].
Die Proteine mit hoher Häufigkeit wie Humanserumalbumin (HSA), Lipoproteine und Globulin werden normalerweise an der Oberfläche von wirkstoffbeladenen NPs adsorbiert, wodurch das in vivo-Freisetzungsverhalten und die Zielorte der NPs verändert werden [10]. Anzahl und Art der adsorbierten Proteine hängen eng mit der Proteinkonzentration im Plasma und der Affinität der NPs zusammen [11]. Je höher die Plasmaproteinkonzentration, desto höher die Oberflächenadsorption des NP [12]. Ein Protein mit hoher Affinität kann das mit schwacher Affinität ersetzen [13]. Daher wird die Oberfläche des NP mit hoher Konzentration und starker Affinität von dem Protein besetzt und bildet eine Nanoproteinkrone [14]. Nanoproteinkronen sind für die in-vivo-Funktion von NPs unverzichtbar [15]. Wenn beispielsweise die Oberfläche mit Polysorbat modifiziert wird, können NPs das Medikament durch die Blut-Hirn-Schranke zum Hirngewebe transportieren [16]; Hydrophob-modifizierte Polysaccharid-Nanomaterialien können mit HSA im Körper interagieren und dadurch die Kontrolle der Wirkstofffreisetzung verbessern [17].
Die Aufnahme von NPs durch Zellen wird durch eine Vielzahl von Faktoren beeinflusst, wie z. B. die physikalischen und chemischen Eigenschaften von NPs, die Konzentration von Nanowirkstoffen, Proteinadsorption und Zelladhäsion [18]. Die Arten und Mengen der Proteinadsorption beeinflussen die Funktion von NPs, einschließlich der Kontrolle und des Targetings der Wirkstofffreisetzung. Auch die physikalischen und chemischen Eigenschaften von NPs, wie Partikelgröße, Ladung und Oberflächenhydrophobie, beeinflussen die Proteinadsorption [19]. Die Natur des NP bestimmt sein Schicksal während des In-vivo-Prozesses [20]. Amphiphile makromolekulare Materialien wie hydrophob modifizierte Polysaccharidpolymere können sich selbst zu Partikeln in Nanogröße zusammenbauen. Die Größe des NP spielt eine wichtige Rolle bei seinen Funktionen des Targetings und der Kontrolle der Wirkstofffreisetzung [21]. Die hydrophobe Gruppe im Polymer ist eine treibende Kraft für die Bildung der Kernstruktur des NP. Je höher der Substitutionsgrad der hydrophoben Gruppe ist, desto kleiner ist der NP [22]. Polymermaterialien mit Carboxylgruppen, Aminogruppen und deren Derivaten sind an der NP-Selbstorganisation beteiligt, sodass sie die Größe der NPs beeinflussen und eine Oberflächenladung bereitstellen, um sich leicht an das Protein mit entgegengesetzter Ladung anlagern zu können [23]. NPs mit unterschiedlichen Oberflächenladungen haben unterschiedliche Fähigkeiten zur Proteinadsorption und unterschiedliche biologische Funktionen [24]. Daher müssen wir die Wechselwirkung zwischen den verschiedenen Oberflächenkomponenten von NPs und Proteinen untersuchen.
HSA ist das am häufigsten vorkommende Protein im Blut. Es ist essenziell für den Transport, die Verteilung und den Stoffwechsel von fremden und körpereigenen Stoffen. Viele niedermolekulare Medikamente gelangen in den Körper und bilden beim Bluttransport HSA-Wirkstoff-Adsorbentien, was die pharmakologische Wirkung von Medikamenten verändert [25]. Wirkstoffbeladene NPs werden nach Eintritt in den Körper mit HSA kombiniert; Aufgrund der komplexen Struktur von NPs unterscheiden sich die Adsorptionseigenschaften von niedermolekularen HSA-Kombinationen [26]. Beispielsweise erfolgt die Adsorption von niedermolekularen Arzneimitteln an HSA-Moleküle schnell; die HSA-Adsorption an NPs ist jedoch langsam und komplex [27].
CHP-Nanopartikel als Wirkstoffträger wurden seit langem untersucht, die ausgezeichnete Nanomaterialien für den Wirkstofftransport gezeigt hatten [28, 29]. In einem früheren Experiment untersuchten wir die Wechselwirkung zwischen HSA und Pullulan-NPs mit unterschiedlichen Graden der Cholesterinsubstitution, cholesterisch hydrophob (CH) modifiziertem Pullulan (CHP), und fanden hauptsächlich zwei Prozesse:HSA heftet sich schnell an die NP-Oberfläche und wird dann langsam in der hydrophobe Kern von NPs [30]. Bei der Bildung von CHP-HSA-Komplexen spielten hydrophobe Wechselwirkungen eine große Rolle [31]. Die Hydrophobie und die Schale-Kern-Struktur der Partikel waren hauptsächlich für die Änderungen der Albumin-Konformation während der NP- und HSA-Wechselwirkung verantwortlich [30].
In dieser Studie stellten wir drei NPs her, CHP, CH-modifiziertes animiertes Pullulan (CHAP) und CH-modifiziertes carboxyliertes Pullulan (CHSP). Ihre Struktur und Eigenschaften wurden mit Fourier-Transform-Infrarot (FTIR) und NMR charakterisiert, und ihre Größen und Potenziale wurden durch dynamische Lichtstreuung (DLS) bestimmt. Isotherme Titrationskalorimetrie (ITC) und Fluoreszenzspektroskopie wurden verwendet, um die Wechselwirkungseigenschaften der NP-HSA-Komplexe und die Auswirkungen der drei Arten von NPs auf die HSA-Struktur zu untersuchen. Wir zeigen die Auswirkungen auf die Wirkstofffreisetzung mit den Eigenschaften der NP-HSA-Komplexe, die für die zukünftige Anwendung von Wirkstoffabgabesystemen von entscheidender Bedeutung sind.
Methoden
Materialien
HSA wurde von Sigma Aldrich (St. Louis, MO, USA) bezogen. N ,N -Imidazol stammte von Shanghai Stock Solution Biotechnology (Shanghai). Ethylendiamin, Bernsteinsäureanhydrid wurde von Tianjin Star Chemical Reagent (Tianjin) bereitgestellt. Alle anderen chemischen Reagenzien waren von analytischer Qualität und stammten von Changsha Huicheng Co. (Changsha, China).
Synthese von CHP, CHSP und CHAP
KWK-Synthese
Cholesterinsuccinat (CHS) wurde wie zuvor beschrieben synthetisiert [32]. Eine Menge von 2 g Pullulanpolysaccharid wurde in 10 ml dehydratisierter Dimethylsulfoxid (DMSO)-Lösung gelöst. Dann wurden 1,06 g CHS, 0,505 g EDC·HCl und 0,268 g DMAP in einer geeigneten Menge DMSO-Lösung gelöst. Die obigen zwei Gruppen von Reagenzien wurden gemischt und bei Raumtemperatur 1 h lang aktiviert, dann in einem erhitzten Ölbad bei 50 °C für 48 h inkubiert. Nach Beendigung der Reaktion und Abkühlen auf Raumtemperatur wurde eine geeignete Menge wasserfreies Ethanol zugegeben und der weiße Feststoff durch Rühren ausgefällt und durch wiederholte Saugfiltration erhalten. Die Produkte wurden mit einer geeigneten Menge wasserfreiem Ethanol, Ethylether und Tetrahydrofuran gewaschen und dann in einem Lufttrockner bei 50 °C getrocknet, um einen weißen Feststoff zu erhalten (Abb. 1).

Synthese von CHP-, CHSP- und CHAP-Polymeren
Synthese von CHAP
Eine Menge von 1,80 g CHP und 1,00 g N ,N -Diimidazol wurden in 100 ml DMSO gelöst. Nach Erhitzen und Rühren in einem Ölbad von 50 °C für 4 Stunden wurden 3,60 g Ethylendiamin zugegeben, gefolgt von weiterem Erhitzen und Rühren für 24 Stunden. Nachdem die Reaktionsflüssigkeit auf Raumtemperatur abgekühlt war, wurde sie in einem 4000-Interception-Dialysebeutel mit doppelt destilliertem Wasser 1 Tag lang dialysiert und dann lyophilisiert, um einen hellgelben Feststoff zu erhalten, der das Produkt von hydrophob modifiziertem animiertem Pullulan war.
Synthese von CHSP
1,80 g CHP wurden in 100 ml dehydratisiertem DMSO gelöst, dann wurden 0,5 g Bernsteinsäureanhydrid und 0,05 g 4-Dimethylaminopyridin (DMAP) in 10 ml DMSO gelöst für 1 h aktiviert; nach 20 h Erhitzen und Rühren in einem 50 °C warmen Ölbad wurde die Reaktion abgebrochen. Als die Reaktionsflüssigkeit auf Raumtemperatur abgekühlt war, wurde sie in eine geeignete Menge wasserfreies Ethanol gegeben und gerührt, um einen weißen Feststoff auszufällen. Der weiße Feststoff wurde mehrere Male mit einer geeigneten Menge wasserfreiem Ethanol, Diethylether und Tetrahydrofuran gewaschen und in einem Gebläsetrockner bei 50 °C getrocknet. Das erhaltene Produkt war hydrophob modifiziertes carboxyliertes Pullulan-Polysaccharid.
FTIR- und NMR-Spektroskopie
Die FTIR-Spektren für CHP, CHSP und CHAP wurden als KBr-Pellets für die FTIR-Spektroskopie erhalten (Nicolet NEXUS 470-ESP, Thermo Fisher Scientific, Waltham, MA, USA). Die chemischen Strukturen von CHP, CHSP und CHAP wurden mit 500 MHz 1 . bestätigt H-NMR, mit DMSO-d6 als Lösungsmittel. Der Substitutionsgrad von Cholesterin in CHP-Polymeren wurde durch die alpha-1,4- und alpha-1,6-glykosidische Bindung und die Methylen-Peakfläche bestimmt.
Vorbereitung und Charakterisierung von NPs
CHP-, CHSP- und CHAP-NPs wurden nach der Dialysemethode hergestellt [33]. Kurz gesagt wurden CHP, CHSP und CHAP in 10 ml DMSO gelöst. Um NPs zu bilden, wurde die Mischlösung 24 h in einen Dialysebeutel injiziert, um das DMSO zu eliminieren. Die Lösung von CHP-, CHSP- und CHAP-NPs wurde mit einem Membranfilter (Porengröße 0,45 m, Millipore, Boston, MA, USA) gescreent, um die größeren aggregierten CHP-, CHSP- und CHAP-NPs zu entfernen. Die Größenverteilung und das Zetapotential der erhaltenen Partikel wurden durch DLS (Zetasizer 3000 HS, Malvern Instruments, Malvern, UK) bei 11,4 V/cm, 13,0 mA bestimmt.
ITC
Auf die CHP-, CHSP- und CHAP NP-Lösungen wurde eine bestimmte Konzentration an HSA-Lösung getropft und die Wärmeänderung mittels ITC (VP-ITC, Microcal, Northampton, MA, USA) gemessen. Eine Menge von 0,9 mM HSA wurde in 0,01 mM CHP-, CHSP- und CHAP NP-Titrationszellen injiziert, um 20-mal zu titrieren. Der erste Tropfen betrug 2 µL und die Reaktionszeit betrug 180 s; die verbleibenden Tropfen betrugen 10 µL pro Tropfen und die Reaktionszeit betrug 210 s und die Temperatur wurde auf 25 °C eingestellt. Die thermodynamischen Parameter und Verbindungskurven wurden mit 28-maliger Titration erhalten.
Fluoreszenzspektroskopie
HSA- und CHP-NPs wurden in einem Molekülverhältnis von HSA zu CHP von 3.6:1 gemischt, um CHP-HSA-, CHSP-HSA- und CHAP-HSA-Mischungen herzustellen. Die erhaltenen Mischungen wurden in 2-ml-EP-Röhrchen gegeben und 24 h bei 20 U/min bei 25 °C geschüttelt. Fluoreszenzspektren und Fluoreszenzintensität (FI) von freiem HSA und NP-gebundenem HSA wurden durch Fluoreszenzspektrophotometrie (Shimadzu RF-4500, Japan) aufgezeichnet. Der Tryptophan-Chromophor im HSA-Molekül wurde bei 280 nm angeregt und Emissionsspektren wurden bei 290 bis 450 nm aufgenommen. Anregungs- und Emissionsspaltbreiten betrugen 5 und 12 nm.
Sieben NP-Lösungen unterschiedlicher Konzentration wurden mit HSA-Lösung gemischt. Die gemischten Lösungen wurden für eine 9-stündige Reaktion in 2-ml-EP-Röhrchen überführt. Die erhaltenen Proben wurden gesammelt, um Fluoreszenzspektren bei einer Wellenlänge von 290–450 nm zu messen. Als Referenz dienten die Fluoreszenzspektren reiner HSA-Lösung zur Bestimmung der Bindungskonstanten nach Stern-Volmer-Analyse. Fluoreszenzlöschungsdaten wurden unter Verwendung der verbesserten Stern-Volmer-Gleichung [34] analysiert:
$$ {F}_0/\left({F}_0-F\right)=1/{f}_{\mathrm{a}}+1/\left({f}_{\mathrm{a}} {K}_{\mathrm{q}}\left[Q\right]\right) $$wo K q ist die Löschkonstante von Stern–Volmer, F 0 und F sind Fluoreszenzintensitäten bei 342 nm in Abwesenheit und Anwesenheit eines Quenchers und [Q ] ist die Konzentration des Quenchers.
Zirkulardichroismus-Analyse
CHP-HSA-Komplexe wurden auf zwei verschiedene Arten hergestellt. Der erste (Komplex I) wurde durch einfaches Mischen von HSA- und CHP-Lösungen hergestellt. Der zweite (Komplex II) wurde in 2-ml-EP-Röhrchen aufbewahrt, die in einem Schütteltisch mit 20 U/min für 12 h bei 25 °C aufgestellt wurden. Circulardichroismus (CD)-Spektren für freies HSA und NPs, die zu Protein hinzugefügt wurden, wurden bei einer Wellenlänge von 200–250 nm unter Verwendung eines CD-Spektrometers (JASCO J-810, Japan) bei 37 °C mit einer 0,1-cm-Küvettenzelle aufgenommen. Die HSA-Konzentration betrug in allen Proben 1,0 mg/ml. Der relative α-Helix-Gehalt in HSA wurde wie folgt berechnet [35]:
$$ \left[{\theta}_{208}\right]=\frac{\theta M}{10 CL{N}_{\mathrm{r}}} $$wo θ 208 ist die mittlere Restelliptizität (Grad cm −2 dmol −1 ) bei 208 nm, θ ist die Elliptizität, M ist das Molekulargewicht von HSA, C ist die HSA-Konzentration (mg/ml), L ist die Länge der Küvettenzelle (cm) und N r ist die Anzahl der Aminosäuren im HSA-Molekül.
In-vitro-Medikamentenfreigabe
Mitoxantron (MTO)-beladene NPs wurden durch eine Dialysemethode hergestellt [36]. Die Standardkurve für Mitoxantron wurde durch UV-Spektrophotometrie erfasst. Wirkstoffbeladung und Verkapselungseffizienz wurden wie beschrieben berechnet [33]. Die MTO-Freisetzung wurde in vitro durch Dialyse in phosphatgepufferter Kochsalzlösung untersucht. Kurz gesagt wurde die Lösung von MTO-beladenen NPs (2 mg/ml) in einen Visking-Dialyseschlauch gegeben und bei 37 °C in einem Luftbad-Schüttler bei 50 U/min gegen das Freisetzungsmedium dialysiert. Zu vordefinierten Zeiten wurde das Freisetzungsmedium gesammelt und das frische Freisetzungsmedium wurde hinzugefügt. Die freigesetzte MTO-Menge wurde durch UV-Spektrophotometrie (UV-384 plus, Molecular Devices, USA) bei 608 nm bestimmt. Der kumulierte Freisetzungsprozentsatz (Q %) wurde wie zuvor beschrieben berechnet [37]. Eine bestimmte Menge HSA-Lösung (0,1 mg/ml) wurde in den Dialyseschlauch gegeben, um die Wirkstofffreisetzung von drei Arten von NPs zu bestimmen.
Ergebnisse
Charakterisierung von CHP-, CHSP- und CHAP-Polymeren
FTIR-Spektren
Abbildung 2 zeigt die FTIR-Spektren für CHP, CHSP und CHAP. Die Daten für die KWK-Spektren waren 1731 cm −1 (–C=O Streckschwingungspeak) und 1161 cm −1 (–C=O Streckschwingungsspitze). Dieses Ergebnis zeigt die Bildung von Esterbindungen auf Pullulan, was darauf hindeutet, dass das CHP erfolgreich synthetisiert wurde.

FTIR-Spektren von CHP (a), CHAP (b) und CHSP (c)
Im Vergleich zu CHP-Spektren betrugen die Daten für CHAP-Spektren 1648 cm −1 (–C=O Schwingungsabsorptionspeak), 1734 cm −1 (–C=O Schwingungsabsorptionspeak), 1539 cm −1 (–N–H Biegeschwingungsspitze) und 3742 cm −1 (–NH2 Dehnungsschwingungsspitze). Gemäß diesen charakteristischen Peaks gab es Amidbindungen an CHP, und CHAP wurde erfolgreich durch die Veresterungsreaktion synthetisiert.
Im Vergleich zu CHP-Spektren betrugen die Daten für CHSP-Spektren 1710 cm −1 (–C=O Streckschwingungsspitze), 1158 cm −1 (–C=O Streckschwingungsspitze), 1560 cm −1 (doppelte –C=O-Kopplungsschwingungsspitze) und 1421 cm −1 (–O–H Biegeschwingungsspitze). Dies zeigt, dass sich an CHP Carboxylgruppen befanden und ein Teil davon zu Salzen wurde.
1 H-NMR
Abbildung 3 zeigt die 1 H-NMR-Spektren für CHP, CHSP und CHAP. Insgesamt 0 bis 2,40 ppm gehörten zum Wasserstoffsignal des Cholesterins, was die erfolgreiche Synthese von CHP belegt. Die charakteristischen Peaks von DMSO-d6 und Methylen (–CH2 CH2 –) zeigten Signale bei 2,49 bzw. 2,53 ppm. Im Vergleich zu CHP zeigte CHAP Signale bei 8–9 ppm, die zur Aminogruppe gehörten und bewies, dass Ethylendiamin auf CHP gepfropft wurde. Der Substitutionsgrad von Cholesterin pro 100 Glucoseeinheiten in CHP könnte aus dem Verhältnis von Methylenprotonen zu Zuckerprotonen mit der folgenden Gleichung berechnet werden [38]:
$$ \mathrm{DS}=\frac{A_{\partial 2.53}}{4\left({A}_{\partial 4.74}+{A}_{\partial 5.01}\right)} $$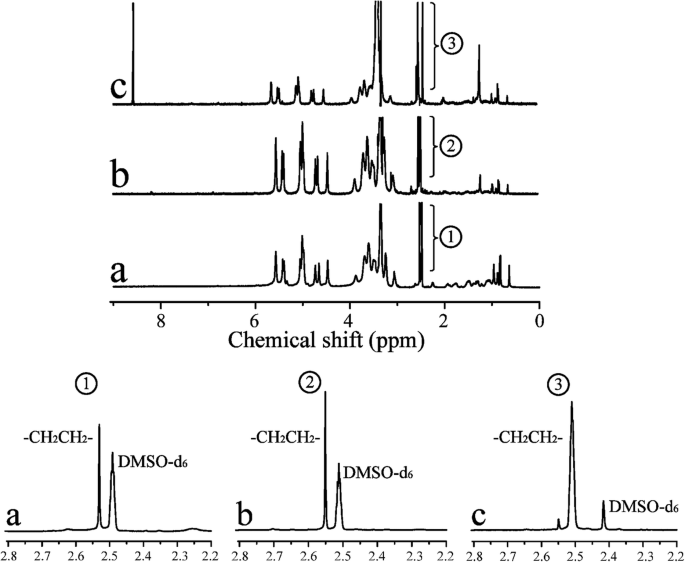
1 HNMR-Spektren für CHP (a), CHSP (b) und CHAP (c) NPs
wo A δ2.53 ist die Spektralfläche unter dem charakteristischen Absorptionspeak von Methylen (Wasserstoff) und A δ4.74 und A δ5.01 sind die Spektralbereiche unter den charakteristischen Absorptionspeaks für alpha-1,6- bzw. alpha-1,4-glykosidische Bindungen.
Von der 1 H-NMR-Spektren in CHP, der Substitutionsgrad von Cholesterinsuccinat (CHS) betrug 4,50%. Bei CHSP-NPs sind die Methylengruppen (–CH2 CH2 –) beinhalteten zwei Aspekte:Bernsteinsäureanhydrid und CHS. Der Substitutionsgrad betrug 12,34 % für Methylengruppen (–CH2 CH2 –) und 7,84% für Carboxylgruppen. Bei CHAP-NPs sind die Methylengruppen (–CH2 CH2 –) umfasste zwei Aspekte:Ethylendiamin und CHS. Der Substitutionsgrad betrug 18,6% für Methylengruppen (–CH2 CH2 –) und 14,1% für die Aminogruppen.
Eigenschaften von CHP, CHSP und CHAP NPs
Amphiphile Polymere können durch Dialyse selbstorganisiert werden, um Kern-Schale-NPs mit einer hydrophilen Hülle und einem hydrophoben Kern zu bilden, die mit Krebsmedikamenten beladen werden können, um wirkstoffbeladene NPs zu bilden. Die NP-Eigenschaften wie Oberflächenladung und Größe spielten einen entscheidenden Einfluss auf die Wirksamkeit der Behandlung als Wirkstoffträger [39, 40]. Die Größenverteilung und das Zetapotential von CHP-, CHSP- und CHAP-NPs, gemessen mit DLS, sind in Abb. 4 dargestellt. Die mittlere Größe der NPs betrug 73,1, 116,9 bzw. 156,9 nm für CHP, CHAP bzw. CHSP. Das Zetapotential betrug − 0,698, + 12,9 bzw. − 15,4 mV (Tabelle 1).

Zetapotential (a ) und Größenverteilung (b ) von KWK-NPs (  ), CHAP-NPs (
), CHAP-NPs (  ) und CHSP-NPs (
) und CHSP-NPs (  )
)
Bei Pullulan-NPs mit derselben hydrophoben Gruppe war die Größe für negativ geladene CHSP und positiv geladene CHAP größer als für neutrale CHP-NPs. Somit stört die geladene Gruppe das Selbstorganisationsverhalten von NPs und bildet in wässriger Lösung größere NPs mit lockerer Struktur. Die Zetapotentiale von CHP-NPs, CHAP- und CHSP-NPs betrugen − 0.698 mV, + 12.9 mV bzw. − 15.4 mV. Somit könnten die Amino- und Carboxylgruppen im Polymer NPs mit unterschiedlichen Oberflächenladungen erzeugen und somit die Oberflächeneigenschaften verändern.
Thermodynamische Analyse
Die thermodynamische Analyse wurde unter Verwendung von ITC mit HSA-Titration auf CHP-, CHSP- und CHAP NP-Lösungen durchgeführt. Als HSA-Titration auf Pullulan-NP-Lösung mit unterschiedlichen Ladungen haben wir die Wärmeänderungen bestimmt und die Verbindungseigenschaften von drei Arten von Materialien, Verbindung und Verbindungsanzahl von Molekülen, ermittelt. Das ursprüngliche Spektrum des isothermen Titrationsthermometers spiegelt die Wärmeänderung wieder. Der nach oben gerichtete Peak weist auf eine wärmefreisetzende Reaktion und der nach unten gerichtete Peak auf eine wärmeabsorbierende Reaktion hin [41, 42]. Wenn CHP-, CHAP- und CHSP-NPs mit HSA titriert wurden, nahm die freigesetzte Wärme aus der Kombination mit der Zeit allmählich ab (Abb. 5). Insgesamt wurden 26 Tropfen HSA-Lösung in CHP NP-Lösung titriert, und die Peaks des Spektrums waren nach oben gerichtet, was auf die exotherme Natur der Reaktion hinweist. Das gleiche Phänomen wurde beobachtet, wenn die HSA-Lösung in die CHSP-NP-Lösung titriert wurde. Wenn jedoch die HSA-Lösung in die CHAP NP-Lösung titriert wurde, waren die Peaks des Spektrums für die ersten vier Tropfen nach oben gerichtet, während sich die Peaks ab dem fünften Tropfen nach unten drehten, was auf eine endotherme Reaktion hinweist. Die HSA-Absorption von CHP- und CHSP-NPs war exotherm, sodass die Reaktion spontan war. Die HSA-Absorption von CHAP-NPs war teilweise endotherm und teilweise exotherm, was mit der positiven Ladung von CHAP zusammenhängen kann.

Isotherme Titrationskalorimetriedaten für die HSA-Titration in a BHKW, b CHSP und c CHAP-NPs bei 25 °C. Die NP-Konzentration in der Zelle (250 µl) betrug 12 µM und die Proteinkonzentration in der Spritze betrug 230 µM. Obere Grafiken zeigen Rohdaten und untere Grafiken zeigen integrierte Vorläufe
Der Enthalpiewert spiegelt die Wärme wider, die durch die Kombination von HSA und NPs freigesetzt wird. Die Enthalpieänderungen von CHP, CHSP und CHAP NPs durch HSA betrugen 42.827, 80.3712 bzw. 22.3951 KJ/mol (Tabelle 2). In Kombination mit der Enthalpieänderung, der exothermen Reaktion, der chemischen Struktur der amphiphilen NPs und der negativen Ladung von HSA kann die hydrophobe Wechselwirkung hauptsächlich die HSA-Wechselwirkung mit CHP- und CHSP-NPs treiben. Bemerkenswert ist, dass bei der HSA-Titration von CHAP die Hitze einen negativen Wert enthielt. HSA-Moleküle enthalten eine hydrophobe Tasche, die mit dem hydrophoben Zentrum des NP kombiniert werden kann [43], sodass die durch die ersten vier Tropfen HSA und CHAP ausgelöste Wechselwirkung hauptsächlich durch hydrophobe Kräfte angetrieben wird. Obwohl HSA negativ und CHAP positiv geladen ist, gibt es ab dem fünften Tropfen aufgrund der endothermen Natur der Reaktion auch eine Ladungskraft zwischen HSA und CHAP.
Der Wert der Entropieänderung kann die Schwierigkeit der Reaktion zwischen HSA und NPs widerspiegeln. Die Entropien von HSA und CHP, CHSP und CHAP-NPs betrugen 0,251, 2,775 bzw. 0,201 KJ/mol K (Tabelle 2), sodass CHP- und CHAP-NPs leichter an HSA zu binden sind, während CHSP-NPs schwieriger zu binden sind an HSA binden.
Die Abdeckung von HSA und CHP, CHSP und CHAP NPs betrug 1,17, 0,404 bzw. 0,845. Die Konzentration der NPs wird basierend auf der Konzentration des Polymers und nicht auf der Anzahl der Partikel berechnet. Wir wissen nicht, wie viele einzelne Polymerpartikel ein einzelnes NP enthalten, daher können wir nicht genau bestimmen, wie viel von jedem NP von HSA adsorbiert wird, aber basierend auf dem Bedeckungswert können wir schlussfolgern, dass CHP-NPs die höchste HSA-Adsorption aufweisen.
In früheren Experimenten haben wir gezeigt, dass der Affinitätswert K A , spiegelt die Stärke der Bindungskraft zwischen NPs und HSA wider [32]. Je stärker die Hydrophobie von CHP-NPs ist, desto stärker ist die Affinität [44]. Die Bindungskonstante von HSA und CHP betrug 27,7 × 10 4 M −1 , die von HSA und CHSP betrug 1,41 × 10 4 M −1 , und der von HSA und CHAP betrug 412 × 10 4 M −1 . Somit war die Kombination aus HSA und positiv geladenem CHAP am stärksten, gefolgt von neutral geladenem CHP und negativ geladenem CHSP. Die stärkste Kombination von HSA und CHAP kann der hydrophoben Kraft zugeschrieben werden, einer gegenseitigen Anziehung der Ladungskraft und wahrscheinlich einer sich gegenseitig ausschließenden Ladung zwischen HSA und CHSP.
Fluoreszenzspektroskopie
Mit Fluoreszenzspektren haben wir die Wechselwirkung zwischen HSA und den drei NPs mit unterschiedlichen Oberflächenladungen untersucht. HSA enthält 585 Aminosäurereste, mit nur einem Trp-Rest bei 214 (Trp214), und sein Fluoreszenzspektrum dominiert im UV-Bereich. Wenn andere Moleküle mit HSA interagieren, kann sich das Fluoreszenzspektrum von Trp abhängig von der Wechselwirkung zwischen HSA und anderen Molekülen ändern. Wenn die drei NPs mit HSA gemischt wurden, erfuhr der maximale Emissionspeak des HSA-Fluoreszenzspektrums keine chemische Verschiebung; nur die Intensität war bis zu einem gewissen Grad abgeschwächt (Abb. 6A). Der maximale Emissionspeak von HSA wurde beobachtet, wenn HSA mit CHAP-NPs kombiniert wurde.

A Fluoreszenzspektren für HSA (1.5 × 10 − 5 mol/L) ohne (a) und mit (b) CHSP, (c) CHP und (d) CHAP NPs mit gleicher Konzentration (4.2 × 10 − 6 mol/l). B HSA-Emissionsintensität mit CHSP-, CHP- und CHAP-NPs bei 343 nm im Zeitverlauf
Experimentelle Studien zeigten, dass die Kombination von HSA und Pullulan-NPs einen komplexierten Prozess aufwies [45]. Wir fügten die drei NPs in HSA-Lösung hinzu und stellten fest, dass die Fluoreszenzintensität der drei NP-HSA-Komplexe allmählich abnahm (Abb. 6B). Das Gleichgewicht wurde für HSA–CHSP bei 556,3 nm nach 12 h, für CHP–HSA bei 534,3 nm nach 10 h und für CHAP–HSA bei 512,3 nm nach 8 h erreicht. Die erforderliche Zeit, um die Fluoreszenzintensität verschiedener NP-HSA-Komplexe auszugleichen, hängt von der vom NP getragenen Ladung ab. Die längste Zeit, die benötigt wurde, um das Gleichgewicht zu erreichen, wurde bei negativ geladenem CHSP-HSA beobachtet, gefolgt von ungeladenem CHP-HSA.
Die schnelle Abnahme der anfänglichen Fluoreszenzintensität der drei NP-HSA-Komplexe (in Abb. 6A gezeigt) ist auf die schnelle Adsorption von NPs und HSA zurückzuführen. Die nachfolgende Fluoreszenzintensität nimmt langsam auf den konstanten Status ab, da die Wechselwirkung von NPs mit HSA ein langsamer Kombinationsprozess ist. Die konstante Fluoreszenzintensität spiegelt den Sättigungszustand der Komplexierung wieder. Die Wechselwirkung zwischen den drei unterschiedlich geladenen NPs und HSA durchlief einen frühen schnellen Rekombinationsprozess und einen späteren langsamen Rekombinationsprozess. Die Kombination von negativ geladenem HSA mit negativ geladenem CHSP erforderte im Vergleich zu ungeladenem CHP eine längere Zeit, um eine komplexe Sättigung zu erreichen. Die Kombination von negativ geladenem HSA mit positiv geladenem CHAP benötigte die kürzeste Zeit, um eine komplexe Sättigung zu erreichen.
Abbildung 7A–C zeigt die Spektren von HSA kombiniert mit unterschiedlichen Konzentrationen von CHSP-, CHP- und CHAP-NPs, um NP-HSA-Komplexe zu bilden. Mit steigender NP-Konzentration nahm der maximale Absorptionspeak des NP-HSA-Komplexes ab, was eine umgekehrte Korrelation zeigt.

Fluoreszenzspektren von HSA (1.5 × 10 − 5 mol/L) mit CHSP (A ), BHKW (B ) und CHAP (C ) bei verschiedenen Konzentrationen (a) 0, (b) 2.07 × 10 −7 , (c) 3,31 × 10 −7 , (d) 4,14 × 10 −7 , (e) 8,28 × 10 −7 , (f) 20,7 × 10 –7 , (g) 33,1 × 10 –7 , und (h) 41,4 × 10 –7 mol/l. D Grundstücke (n = 7) für F 0 /(F 0 − F) vs 1/[Q ], Q ist die Konzentration von CHSP (–◆–), CHP (–■–) bzw. CHAP (–▲–)
Das positiv geladene CHAP-HSA zeigte die kürzeste Zeit, um das Gleichgewicht zu erreichen.
Wir haben eine modifizierte Stern-Volmer-Gleichung verwendet, um die Fluoreszenzlöschungsdaten zu analysieren:
$$ {F}_0/\left({F}_0-F\right)=1/{f}_{\mathrm{a}}+1/\left({f}_{\mathrm{a}} {K}_{\mathrm{q}}\left[Q\right]\right) $$wo f a ist der Kontaktanteil der fluoreszierenden Substanz mit dem Quencher, K q ist die Stern-Volmer-Löschkonstante, F 0 ist die Fluoreszenzintensität bei 342 nm ohne Quencher, F die Fluoreszenzintensität bei 342 nm mit dem Quencher und [Q ] ist die Konzentration des Quenchers.
Aus dem Funktionsbild von F 0 /(F 0 − F ) Paar 1/[Q ] erhalten wir die Werte von f a und K q von der Steigung und dem Schnittpunkt (Fig. 7D). Unter der Annahme, dass sich die beobachtete Fluoreszenzintensität hauptsächlich durch die Wechselwirkung zwischen NPs und HSA ändert, kann die Löschkonstante als Bindungskonstante für die Komplexbildung angesehen werden. The binding constants for HSA and CHSP, CHP, and CHAP molecules were 2.02, 2.99, 4.72 × 10 5 M −1 , bzw. In the previous study, we discussed the interaction between HSA and CHP NPs with different hydrophobicity substitutions [30]. The hydrophobic interaction between HSA molecules and CHP cholesterol played an important role in the formation of CHP–HSA. The greater the hydrophobic substitution of CHP, the greater the binding constant of CHP and HSA.
In the present study, we found that the value of the binding constant (K b ) is related to the electrical properties of the NP. A surface with a positive charge, such as CHAP, has the largest K b , and a surface without a charge, such as CHP, has the second largest K b . The K b for CHSP, with a negative charge, was the lowest. Therefore, in addition to the hydrophobic interaction between NPs and HSA, the electrostatic interaction between them also plays an essential part in the formation of NP–HSA complexes.
In addition, the f a values for CHSP, CHP, and CHAP NPs were 0.269, 0.288, 0.38, respectively, so part of the Trp residue was involved in this reaction. Furthermore, the amount of HSA was too much at a given NP/HSA concentration, so free HSA molecules presented in the reaction system.
CD Spectrum Analysis
Figure 8A shows the CD spectra (a) free HSA, (b) CHSP–HSA, (c) CHAP–HSA, and (d) CHP–HSA in solution at 25 °C. The samples were complex I. There are two negative bands at 208 and 222 nm of the UV region in HSA spectra, which are the characteristic peaks of the α -helical structure. Die α -helical content of free HSA was 55%. At the beginning of the complex, with the recombination of HSA and CHSP, CHAP, and CHP NPs, the α -helical content of HSA was reduced to 52.0%, 48.57%, and 48.0%, respectively.

A CD spectra for (a) free HSA, (b) CHSP–HSA, (c) CHAP–HSA, and (d) CHP–HSA in solution at 25 °C. The samples were complex I. B CD spectra for (a) free HSA, (b) CHSP–HSA, (c) CHAP–HSA, and (d) CHP–HSA in solution at 25 °C. The samples were complex II. C Ellipticity at 208 nm for HSA interacting with CHSP (–▲–), CHAP (–●–) and CHP (–■–) over time
Figure 8C shows that the ellipticity changes of the three samples at 208 nm over time. The ellipticity of HSA combined with CHAP and CHP NPs increased from 0 to 12 h and leveled off at 12 h, but that of HSA with CHSP NPs increased faster, i.e., the ellipticity increased from 0 to 9 h and leveled off at 9 h. Therefore, the ellipticity of the three samples gradually increased over time and the α-helical content gradually decreased; when the ellipticity maintained constant, the recombination of the sample and HSA was completed.
Figure 8C illustrates that the fastest surface absorption rate was presented on the combination of CHSP and HSA. The size, charge, and hydrophobicity of NPs can influence the migration rate of HSA to the center of NPs. The surface of CHP is not charged, the surface of CHSP is negatively charged, and the surface of CHAP is positively charged. Owing to the presence of the negative-charge mutual exclusion between CHSP and HSA, the resistance of HSA migrating to the CHSP NP center becomes larger. The traction of NPs on HSA is driven by the hydrophobic interaction forces. The traction of the three NPs on HSA is identical, so the lowest velocity of HSA migrating to the center of NPs was observed on the combination of HSA and CHSP NPs. Because the particle size is in the order of CHSP> CHAP> CHP, the particle density displays a reverse order, i.e., CHSP
The ITC results show that the amount of HSA migrating toward the center of CHSP NPs is the smallest but with the fastest speed compared with other types of NPs. Table 3 shows that the α-helix content of HSA migrating toward the center of CHP NPs was the lowest. The more the secondary structure of HSA was damaged, the faster the HSA migrated toward the center. The fastest speed was observed on HSA migrating to the center of CHSP NPs (Fig. 8C).
Figure 8B shows CD spectra for (a) free HSA, (b) CHSP–HSA, (c) CHAP–HSA, and (d) CHP–HSA in solution at 25 °C. The samples were complex II. After the complexation is completed, the α -helical content of HSA recombined with CHSP, CHAP, and CHP was reduced to 46.27%, 44.55%, and 42.91%, respectively (Table 3). With the increase in interaction time, the secondary structure of HSA was changed and the α -helical content was reduced during the process of complexation with NPs.
We measured the drug release rate of MTO with the three kinds of drug-loaded NPs and drug-loaded NP–HSA complexes. The drug release for free MTO was about 99.8% at 4 h (Fig. 9). The release rate in 48 h for CHP, CHAP, and CHSP NPs was 53.68%, 58.54%, and 63.24%, respectively. The drug release from all NPs was fast in the first 8 h, which was a burst release process, and the drug release remained stable after 12 h, which was a sustained release process. In vitro drug release from NPs is not affected by gastrointestinal pH and enzymes, and the dissolution of nanomaterials results in little drug release. The release is mainly determined by dissolution and diffusion [46]. The 48-h drug release rate was in the order of CHSP> CHAP> CHP, corresponding to the size of NPs. The fastest release rate was found on CHSP, which was negatively charged with the largest size. The second fast drug release rate was found on which was positively charged with the size smaller than CHSP. CHP was electrically neutral, and its drug release rate was minimal. Hence, the polymer surface groups involved in the formation of NPs affect the size of NPs and ultimately the drug release of NPs.
Mitoxantrone (MTO) release of pullulan NPs in phosphate-buffered saline (PBS) at 37 °C in vitro (□:free mitoxantrone, ○:CHP, △:CHAP, ▽:CHSP, ◁:CHAP–HSA,◇:CHP–HSA, ▷:CHSP–HSA)
The drug release rate from the combinations of HSA and CHAP, CHP, and CHSP in 48 h was 32.45%, 33.86%, and 35.76%, respectively. After the combination of NPs and HSA, the drug release in 48 h was significantly decreased as compared with HSA-free NPs, which was mainly attributed to the resistive effect and adsorption effect of HSA. At 48 h, the drug release of the compounds was in the order of CHSP–HSA> CHP–HSA> CHAP–HSA, while the drug release of HSA-free NPs was in the order of CHSP> CHAP> CHP. Although NP–HSA compounds showed significantly slow drug release, the total release of CHP-HSA decreased by 21.23% in 48 h, whereas the total release of CHAP-HSA decreased by 25.68% and that of CHSP-HSA by 28.48%. The drug release of the NPs is related to the size of the NPs and the polymer hydrophobic groups on NP surface. The adsorption of HSA can lead to significant slowdown in drug release, which is related to the hydrophobicity of NPs and also to the surface charge of NPs [46]. The adsorption of HSA is closely related to the size of the NPs and the degree of substitution of the hydrophobic groups of the polymer during the self-assembly process. Nevertheless, the drug release of the NPs is ultimately determined by the properties of the NP itself.Drug Release

Discussion
As shown in Fig. 10, the formation of the NP–HSA complex is driven by a hydrophobic force between cholesterol groups of the particle core and the aromatic amino acid of the hydrophobic domain of HSA. After mixing, HSA interacts with the surface cholesterol unit and is rapidly adsorbed to the NP surface. Then, the adsorbed HSA on the NP surface is processed because of the hydrophobic forces derived from the cholesteric unit in the particle core. When overcoming the steric hindrance of polysaccharide chains in the NP shell, the adsorbed HSA gradually migrates to the core. After the hydrophobic interaction and resistance of the hydrophilic polysaccharide chain are balanced, the HSA molecule enters the particle core to become hydrophobically bound to cholesterol groups to form the NP–HSA complex.

Adsorption of HSA to NPs
For CHAP and CHSP NPs, the recombination of HSA is a complex process also subjected to the charge interaction with HSA under the traction of the hydrophobic driving force. The binding constants of the three kinds of NPs with the same hydrophobic substitution and different surface charge were in the order of CHAP> CHP> CHSP. The electrical properties also play a major part in the formation of NP–HSA. In this process, the formation of the CHSP–HSA complex was blocked by the structure of the NP shell and the repulsive force between the negative charges, which led to their loose connection. During the rapid adsorption and slow recombination, the degree of spiraling of HSA is lower for CHSP NPs than CHP NPs and CHAP NPs. Therefore, the surface charge of NPs not only changes the nature of the particles themselves but also affects the protein complex.
In the current study, we investigated the effect of NP surface charge on the interaction between NPs and proteins (Fig. 10). Three different charges of pullulan NPs with HSA adsorption still showed rapid adsorption and slow recombination. The number of HSA molecules with positively charged CHAP complex was the most, including rapid adsorption of NP–HSA by hydrophobic forces, HSA molecule migration to the center, and the HSA molecules adsorbed on the surface of NPs by charge action. CHP- and CHSP-adsorbed HSA molecules were mainly distributed in the hydrophobic center of NPs, with CHSP adsorbing fewer HSA molecules. The adsorbed number of HSA molecules is related to the hydrophobicity of NPs. The greater the degree of substitution of hydrophobicity, the more HSA is adsorbed [41]. The cholesterol substitutions of the three NPs were the same, and the number of HSA molecules adsorbed by positively charged NPs was the highest, so the adsorption of NPs and HSA was related to the hydrophobicity and surface charge of the NPs.
The surface adsorption capacity between NPs and HSA is also related to the hydrophobicity and charge of NPs. The binding force between HSA and NPs is determined by the hydrophobicity, surface charge, size, and structure of NPs. The α-helicity was decreased most at the beginning of adsorption and the complete CHP–HSA complex. CHP NP has the smallest size and highest density. The CHP NPs migrated toward the center by the hydrophobic traction; the sugar chain of the CHP NP shell was larger to inhibit the migration toward the center. The extension of the peptide chain of HSA is larger, with the α -helix decreased the most. Although CHAP NPs have hydrophobic and charge forces, they possess relatively large size, loose structure, small resistance in the periphery, small extension of the peptide chain, and small content of the α -helix. Some HSAs remained on the surface of NPs through the charge force of adsorbing, and the α -helical content is also smaller in this part of the HSA. Die α -helix content of CHAP decreased less than that of CHP, mainly due to the peptide chain extension-induced central pulling force which led to α-helix content decline. During the process of the CHSP and HSA complexation, the role of the central pulling force has a reverse direction of the charge force, thereby resulting in weakening the center of the migration force. CHSP NPs are larger than CHAP NPs, and the structure of CHAP NPs is loose. Because the adsorbed number of HSA on CHAP is higher than that on CHSP, the decrease of α -helicity in CHSP is less than that in CHAP NPs. Therefore, the interaction between NPs and HSA and the decrease in α -helicity are all related to the size, density, hydrophobicity of substitution, surface charge of the NPs, and number of HSA connections.
After the NPs enter into the blood, protein adsorption affects the functions of NPs, such as the slow and controlled drug release, the travel from the blood circulation passing through the vascular barrier, targeting tissue, and entering cells. NPs interact with the HSA in the body and affect the in vivo behavior of NPs. The number of adsorbed proteins is closely related to the properties of the NPs. HSA adsorbs NPs, which affects the distribution in organs and removal of NPs, thereby altering the concentration of the drug in the body and the efficacy of the drug.
Finally, the properties of NPs, such as size, hydrophobicity, and surface charge, affect the drug release of NPs in vivo. We can design specific materials to perform specific functions with specific protein adsorption.
Conclusions
In this study, three kinds of nano-drug carriers were constructed, CHP, CHSP, and CHAP. The size, charge, drug loading properties of NPs, interaction between NPs and HSA, and drug release were all closely related to charge amount and charge type of nanomaterials. With the same degree of substitution of hydrophobicity, CHAP NPs with larger amino substitutions were the largest, CHSP NPs the second largest, and CHP NPs the smallest. The size and surface charge of the NPs were essential to the coverage of HSA, the binding constant, and the slow drug release. The positively charged CHAP binding constant was the strongest, showing the fastest drug release, and CHP NPs had the highest coverage. The combination of HSA further retarded the drug release of NPs. CHAP NPs adsorbed HSA had the slowest drug release rate.
Abkürzungen
- CH:
-
Cholesteric hydrophobically
- CHAP:
-
CH-modified animated pullulan
- CHP:
-
Cholesteric hydrophobically (CH) modified pullulan
- CHSP:
-
CH-modified carboxylated pullulan
- DMSO:
-
Dehydrated dimethyl sulfoxide
- HSA:
-
Human serum albumin
- K b :
-
The binding constant
- MTO:
-
Mitoxantrone
- NP:
-
Nanoparticle
Nanomaterialien
- Komplexe Spannungs- und Stromberechnungen
- Nanofasern und Filamente für eine verbesserte Wirkstoffabgabe
- Biokompatible FePO4-Nanopartikel:Wirkstofftransport, RNA-Stabilisierung und funktionelle Aktivität
- Abstimmung der Oberflächenchemie von Polyetheretherketon durch Goldbeschichtung und Plasmabehandlung
- Bildung und Lumineszenzeigenschaften von Al2O3:SiOC-Nanokompositen auf der Basis von durch Phenyltrimethoxysilan modifizierten Aluminiumoxid-Nanopartikeln
- Abstimmung der Oberflächenmorphologien und -eigenschaften von ZnO-Filmen durch das Design der Grenzflächenschicht
- Elektrospinnen auf isolierende Substrate durch Kontrolle der Oberflächenbenetzbarkeit und -feuchtigkeit
- Die Kopplungseffekte von Oberflächenplasmonpolaritonen und magnetischen Dipolresonanzen in Metamaterialien
- 5-Aminolävulinsäure-Squalen-Nanoanordnungen für die Tumorphotodetektion und -therapie:In-vitro-Studien
- Einfluss der elastischen Steifigkeit und Oberflächenhaftung auf das Prellen von Nanopartikeln